




BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
URL: http://ijmm.ir/article-1-2546-en.html



2- Department of Biology, College of Science, University of Misan, Maysan, Iraq ,
3- Department of Plant Protection, College of Agriculture, University of Misan, Maysan, Iraq
The faba bean (Vicia faba L.), also known as the broad bean, is a widely cultivated leguminous crop with a long history of agricultural significance. Native to the Nile Valley, North Africa, and Central Asia, it has remarkably adapted to diverse environmental conditions worldwide (1, 2). Valued for its high protein content (20–30%) and rich energy profile, faba bean is a vital food source for humans and livestock (3, 4). Additionally, it plays a crucial role in nitrogen fixation through symbiotic interactions with root-associated rhizobia, enhancing soil fertility and sustainability (5). Given its agronomic and ecological benefits, faba bean holds significant potential for improving soil health and supporting sustainable agricultural systems (6).
Root rot disease is one of the major biotic factors that infect broad bean crops and is responsible for severe productivity loss at 5–30% and even reach to 100% under its suitable environmental circumstances (7, 8). Infection of the roots by the soil borne pathogens can affect water and nutrient uptake (2, 9). Fungal diseases are commonly caused by multiple soil borne fungi, including Fusarium spp., Rhizoctonia spp., Pythium spp., phanomyces spp., and Phoma spp. (10, 11). Several fungi can infect plants through the root system, although stem bases, crowns, and seedlings are their primary targets (12, 13).
Different standard methods have been used to identify the fungi species. Morphological analysis, both macroscopic and microscopic observations, are the primary identification methods (14). Molecular identification for the accurate recognition of fungi is an alternative to morphological analysis. Moreover, molecular sequences are used for accurate identification to identify and differentiate some species showing the same morphological features (15). Thus, molecular methods have become broadly used for the taxonomy of species (16), which is important to identify the Internal Transcribed Spacer (ITS) of the fungi species that play a major role in the phylogenetic classification of genera, as its level is determinant even among the isolates of the same species (17). The ITS area is among the sings with the highest probability of correct recognition for a very wide group of sampled fungi. More fungal studies have supported the ITS region as an appropriate barcode of fungi (18).
ITS and rRNA genes have been used for fungal classification over the last two decades. The fungal DNA sequence for the 28S and 18S subunits and the ITS is related to the fungal identification (19). DNA forensic method uses two internal transcribers (ITS1 and ITS2) regions of the nuclear rDNA to estimate the polymorphisms (20). The ITS areas are suitable for the identification of the fungal species because of their long sequential polymorphisms. DNA sequence analysis of ITS1 and ITS2 is common and efficiently-used method for the fungal identification (21).
The sequences were compared and the BLAST analysis was done with NCBI data with coordinated agreement sequence (22). Moreover, the multiple alignment software ClustalW was used to match the highest similarity degree query coverage with the best maximum identical 10 sequences (17).
CLUSTALW was used to determinate and align with the closest species, in addition to detection of geographical variations (16).
Broad beans are favorite food for the people of Iraq and are consumed in large quantities. Therefore, they are cultivated in different areas due to the abundance of water, fertile soil, and suitable available environmental conditions. The principal aims of this study were to identify 8 species of the root rot fungi isolated from the root of broad bean fields in Maysan Governorate/Southern Iraq using molecular study.
2.1 Sampling
The species were obtained from the Fungi Laboratory, Department of Biology, College of Science, University of Misan, Iraq. Eight species of root rot fungi were isolated from the roots of broad beans grown in the fields in Maysan Governorate. The strains were selected phenotypically and identified through DNA extraction and PCR assays.
2.2 DNA Extraction and Amplification
The isolated fungi were grown on potato dextrose agar (PDA) for 7 days at 22–25°C. Genomic DNA was extracted from eight isolates of mycelium samples using the Fungi Genomic DNA Extraction Kit (Presto™ Mini gDNA Yeast Kit/ Genaid/ Taiwan). The amplification was performed using master mix, primers, DNA samples, and water to the volume of 25 µl. The cycling program was as follow: initial denaturation at 95°C for 5 min, 35 cycle of denaturation at 95°C for 0.03 sec, annealing at 56°C for 0.03 sec, and extension at 72°C for 1 min, and 1 cycle of final extension at 72°C for 10 min.
2.3 PCR-RFLP
The samples were sent to the Macrogen company/ South Korea (https://www.macrogen.com/en/main) to identify DNA sequences. Sequences of the tested species were registered in the GenBank database and each isolate received an accession number (Table 1). The information obtained from the DNA sequences were compared with the sequences of other fungal isolates available in National Center for Biotechnology Information (NCBI) GenBank database. Blast search was used to diagnose species and the closest matched sequences were obtained. Moreover, phylogenetic trees were created using the Molecular Evolutionary Genetic Analysis (MEGA) software version 11.0 (23).
The majority of morphological characteristics are highly differentiated with no specificity. The phylogenetic analysis results of the sequence of nitrogenous bases established the genetic relationship between fungal isolates identification in this study with a group of fungal isolates in the world of the identical genus and species. To evaluate the genetic varieties and relationship between the tested fungi, molecular identification was performed using PCR for the isolates, using the ITS1 and ITS4 primers to amplify the ITS area. All tested fungi produced fragments of 500–700 bp. The accession numbers of a total of 8 strains of the root rot fungi, identified in Maysan Governorate, Southern Iraq, were registered in NCBI GenBank. This study showed that the sequences of all tested fungi were 98.40 to 100% identical to the sequences of the reference strains in the GenBank (Table 1).
Table 1. The phenotypic, molecular identification, and references to isolated fungi strains in GenBank.
| Identical % | GenBank sequence accession number | Number of reference | Molecular identification | Phenotypic identification |
| 99.05 | LC818109 | MK919294.1 | C. sepedonium | Corynascus sepedonium |
| 99.67 | LC818113 | ON803511.1 | F. oxysporum | Fusarium oxysporum |
| 99.97 | LC818106 | MT509567.1 | F. solani | F. solani |
| 98.48 | LC818112 | MT127399.1 | M. phaseolina | Macrophomina phaseolina |
| 100 | LC818110 | MT319766.1 | M. fragilis | Mucor fragilis |
| 100 | LC818111 | MT077159.1 | M. inundatum | Myrothecium inundatum |
| 98.90 | LC818108 | MW829525.1 | O. pseudomollicella | Ovatospora pseudomollicella |
| 98.40 | LC818107 | KF907736.1 | R. solani | Rhizoctonia solani |
The strains M. inundatum and M. fragilis showed 100% identity with GenBank sequences and C. sepedonium, F. oxysporum and F. solani showed more than 99% identity. Our strain C. sepedonium which was given the accession number LC818109, showed 100% identity with reference strain MZ203485.1, while MH664052.1, MK883679.1 and KP721604.1 reference strains were genetically far (Figure 1). F. oxysporum (accession number LC818113) indicated 100% similarity with many isolates previously registered in NCBI such as OP962344.1, OR879163.1 and OR879157.1 (Figure 2). Figure 3 represents phylogenetic tree of F. solani (LC818106), which showed similarity (100%) to MN857747.1 and MT509567.1 strains.
On the other hand, Figure 4 shows MT127390.1 as the closest reference strain to M. phaseolina (LC818112), while, M. fragilis (LC818110) showed 100% identity with many reference strains (FJ904925.1, MT319766 and MT319765) (Figure 5). M. inundatum (LC818111) revealed 100% similarity with both OP761606.1 and MK024176.1 strains (Figure 6), on the other hand, O. pseudomollicella (LC818108) indicated 100% similarity with MW251835.1 and MW242833.1 reference strains (Figure 7). Finally, the strain R. solani, which was given the code LC818107, showed 100% similarity with many strains such as AF153786, MH014991.1, and AF153788.1 (Figure 8).
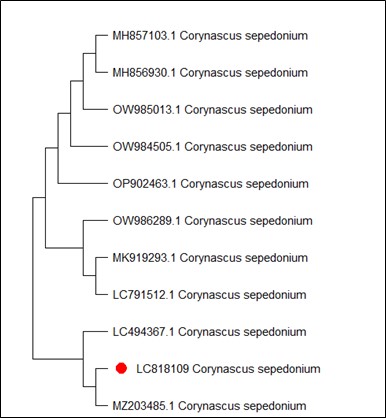
Figure 1. Phylogenetic tree according to the sequencing results of Corynascus sepedonium

Figure 2. Phylogenetic tree according to the sequencing results of Fusarium oxysporum

Figure 3. Phylogenetic tree according to the sequencing results of F. solani
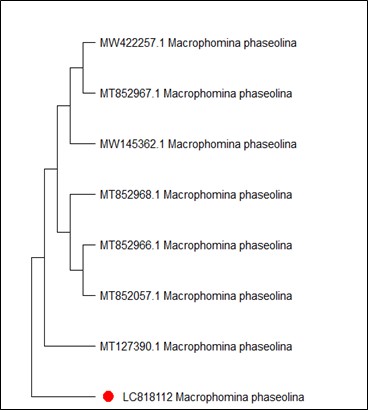
Figure 4. Phylogenetic tree according to the sequencing results of Macrophomina phaseolina

Figure 5. Phylogenetic tree according to the sequencing results of Mucor fragilis
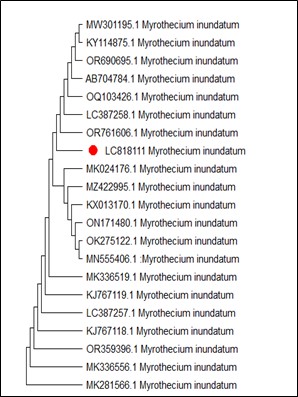
Figure 6. Phylogenetic tree according to the sequencing results of Myrothecium inundatum
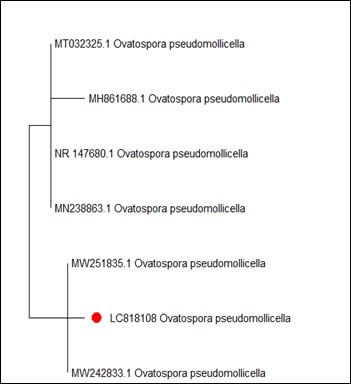
Figure 7. Phylogenetic tree according to the sequencing results of Ovatospora pseudomollicella

Figure 8. Phylogenetic tree according to the sequencing results of Rhizoctonia solani
Faba bean is the common fast food in Middle East countries diet such as Iraq. It is a good source of carbohydrate, protein, dietary fiber, lecithin, choline, and secondary metabolites (24). Broad bean root rot is a serious disease caused by many pathogenic fungi, including Rhizoctonia Solani, Fusarium spp., Macrophomina phaseolina, and Pythium spp. (25).
Proper diagnosis of the fungal pathogens in plants is considered as a mean of understanding for the relationship between pathogenic fungi and plants in agricultural environment (26) for the purpose of preparing strategies to control fungal diseases whether they are specific or complex pathogens. Phenotypic and molecular characteristics provide accurate and complete information of the root pathogens (27).
Eight species were isolated from the root rot of faba bean. They were identified morphologically and using molecular analysis. Furthermore, the isolates of species from this study were recognized at species level by sequencing of the ITS rDNA region and then submitted to BLAST for the similarity search. These sequences were compared with other reference sequences of the most common species reported in GenBank database.
It was shown that the morphological identification completely matched to the molecular identification with 98.40 to 100% similarity with the sequences of the reference strains in the GenBank.
Corynascus sepedonium has been isolated from many habitats such as rhizoplane of plants and marine soils (28). Our C. sepedonium isolate showed 100% identity to the reference strain belong to the genus Corynascus, while, Atashi Khalilabad and Fotouhifar (29) showed 98-99% similarity between many isolates of C. sepedonium (MSRT-R5, MK919294.1) and C. verrucosus (MN956890.1), forming sister branches in phylogenetic tree, whereas other isolates showed 83% bootstrap value from a distinct clade.
Identification of F. oxysporum and F. solani based on the phenotypic properties needs lots of experience in Fusarium taxonomy (30). Therefore, application of the advanced, accurate and rapid assays are required to identify Fusarium species. PCR techniques are the most important methods for the molecular determination of the plant pathogenic fungi (31). F. oxysporum and F. solani are important pathogenic fungi on several economically important crops, causing Fusarium root rots (32). Molecular analysis of both fungi indicated the presence of broad genetic differences among the isolates. Further analysis discovered intra-species differentiation and nucleotide addition or deletion in the ITS area among the isolates (33, 34).
The M. phaseolina isolate was found to be identical (100%) to the reference strains. While, Mnati et al (34) showed that their isolates had a genetic difference, ranging between 94%-99%, with the several isolates previously registered in the GenBank. Our isolate was registered in NCBI under the accession number LC818112.
Khan and Javaid (35) revealed that most of the isolates of M. fragilis were identical to the NCBI reference strains. Our strain of M. fragilis isolate was also identical to the most of the isolates preserved in the GenBank, which was in accordance with Khan and Javaid (35) study. Moreover, molecular studies are accurate techniques to differentiate between M. fragilis and other species (36), especially M. variicolumellatus (37).
Myrothecium comprises several species such as M. inundatum (38). Identification of Myrothecium species is difficult due to the few distinct morphological features and the absence of voucher samples with molecular information (39). Therefore, molecular studies of more isolates of M. inundatum are very necessary to remove doubts about its taxonomy.
The genus Ovatospora belonging to the Chaetomiaceae family contains nine species (40). Ovatospora pseudomollicella was recorded for the first time in Iraq by Alhussani et al (41). It has been shown that many species of Ovatospora are most similar to the species of Chaetomium (42).
Our R. solani isolate showed 100% identity to many GenBank strains. This result did not agree with Al-Fadhal et al (43) study who indicated their R. solani isolate differed from R. solani GenBank isolates. While Dakhil et al (44) represented R. solani isolates recovered from Iraqi soil with identify to the ones registered in GenBank.
Identification of fungal species based on morphological features requires great expertise in taxonomy of fungi. Therefore, development of a rapid, accurate, and specific procedure for identification of species is required. The use of methods based on nucleotide sequences including PCR are the most important equipment for molecular diagnosis of pathogenic fungi (14, 31). The evidence of taxonomic morphology are confirmed by PCR analysis (45).
Several studies showed that PCR method and restriction enzymes can be used for the accurate classification of several pathogens such as root rot fungi (46, 47). Moreover, universal primers are used for identification and taxonomy of fungi. Use of ITS1 and ITS2 regions, which are surrounded by 5.8 SrDNA gene (29, 47) are considered convenient to identify the fungal species that are ecologically important. Identification of the fungi species is possible using Sanger sequencing and doing BLAST of the sequence in wide GenBank sequence database (48).
This study highlighted high degree of identity of the fungal isolates from Iraqi soil with the isolates preserved in the GenBank, with a similarity percentage reaching more than 99%. Moreover, the use of modern molecular methods in diagnosing fungi and the use of diagnostic primers to study other genes helps us obtain a better classification, especially at the species level.
The authors extend their appreciation to the College of Science, University of Misan for funding this study. Authors are heartily thankful to Asst. Prof. Dr. Maitham Dragh, Head of the Department of Biology for his sincere assistance in completing the research.
Ethical Considerations
Not applicable.
Authors’ Contributions
Ali A Kasim and Ghassan Mahdi Daghir (supervisors): Developed the research idea, designed the methodology, directed data collection, reviewed the manuscript, enhanced the discussion with modern references, and arranged and edited the research and wrote it in English, ensured linguistic and scientific quality, and prepared the final draft for publication. Asia N Kadim collected samples, conducted all laboratory experiments, wrote the results, discussed and wrote the references. All authors have read and approved the final version of the manuscript. The final manuscript was read and approved by all of the authors.
Conflicts of Interest
There is no grant support or financial relationship associated with this study. The master’s student paid most of the research costs.
The authors were not utilized AI Tools.
Received: 2024/12/12 | Accepted: 2025/02/18 | ePublished: 2025/03/30
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
Copyright Policy
Iranian Journal of Medical Microbiology by Farname is licensed under CC BY-NC 4.0![]()
![]()
![]()
