




![]()
![]()
![]()
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
URL: http://ijmm.ir/article-1-1395-en.html


 , Golshid Javdani Shahedin2 , Ahmad Nejati3 , Iradj Ashrafi4 , Mahla Asadian5 , Ramin Mazaheri Nezhad Fard6
, Golshid Javdani Shahedin2 , Ahmad Nejati3 , Iradj Ashrafi4 , Mahla Asadian5 , Ramin Mazaheri Nezhad Fard6
2- Pasteur Institute, Tehran, Iran
3- Department of Virology, School of Public Health, Tehran University of Medical Sciences, Tehran, Iran
4- Faculty of Veterinary Medicine, University of Tehran, Tehran, Iran
5- Department of Pathobiology, School of Public Health, Tehran University of Medical Sciences, Tehran, Iran
6- Department of Pathobiology, School of Public Health, Tehran University of Medical Sciences, Tehran, Iran ,
Enterococci are Gram-positive catalase-negative bacteria that are naturally present in the gastro-intestinal tract (GIT) of humans, animals, and the environment. Enterococci were reported to be the leading cause of antibiotic-resistant infections in the human bloodstream, urinary tract (UI), and surgical wounds in the 1970s and 1980s. Antibiotic resistance is a major threat to human health worldwide and significantly increases health care costs (1). Two important and pathogenic species of enterococci are E. faecium and E. faecalis. Enterococcus faecium as a human pathogen is particularly important due to its high resistance to various antimicrobial drugs such as ampicillin and vancomycin (2). In general, up to 80% of E. faecium isolates in hospitals are resistant to vancomycin (3). The rapid spread of vancomycin resistance in these strains greatly challenges the ability of physicians to treat infections as no other antimicrobial drugs are often available (4).
Accurate identification of vancomycin-resistant enterococci (VRE) reservoirs and transmission path-ways is critical for strategic infection control interv-entions (5, 6). For example, enterococci could transmit vancomycin resistance to methicillin-resistant Staphy-lococcus aureus (MRSA) (7). Whole-genome sequenc-ing (WGS) is a comprehensive high-resolution method used to sequence a wide range of bacterial species and mapping of pathogen trans-mission routes (8). Enterococcus faecium genetic features have been investigated specifically to identify the bacterial lineages (9). Factors associated with E. faecium infections in humans include virulence factors (VF), antibiotic resistance (AR) genes, mobile genetic elements (MGE), and multiple-locus sequencing (MLST) patterns (10, 11). The aim of this study was to sequence and analyze the multidrug-resistant E. faecium EntfacYE genome, focusing on the genes involved in antibiotic resistance.
Isolation of E. faecium EntfacYE
Twenty-five clinical enterococci were isolated from patients' blood from 2019 to 2020. Sampling was carried out in a university teaching hospital, Tehran, Iran (ethics approval no. IR.TUMS.SPH.REC.1397.139). Of all the enterococcal isolates, one E. faecium strain isolated from a clinical blood sample that three various bacteriophages were isolated on it, was selected in this study (E. faecium EntfacYE) (12). The bacterial strain was cultured on trypticase soy agar (TSA) media for purification. Furthermore, the isolate was verified using morphological, biochemical, and molecular techniques. Assays such as Gram staining, catalase, growth in 6.5% NaCl, and glucose utilization were used for the isolate as well.
Furthermore, the isolates were assessed for resistance to common antibiotics using the disk diff-usion method. The isolate was cultured on Mueller-Hinton agar media and exposed to the selected antibiotics at 37°C for 24 h. The assessed antibiotics included cefoxitin, ceftriaxone, clindamycin, erythr-omycin, linezolid, and vancomycin. After incubation, inhibition zone diameters were measured, and a bacterial resistance pattern was reported.
Sanger Partial Sequencing
Molecular identification of the isolate was carried out using PCR amplification of the elongation factor Tu (EF-Tu) gene. Then, a PCR amplicon was sequenced using the Sanger method (Kawsar Biotech, Iran). Briefly, a single bacterial colony was dissolved in sterile distilled water (DW) in a sterile microtube. To extract the bacterial genome, the microtube was incubated at 90°C for 30 min and then centrifuged at 7500 g for 5 min. The genome concentration was measured, and the ratios of 260/280 and 260/230 nm were calculated using NanoDrop One (Thermo Fisher Scientific, USA). Then, the extracted bacterial genome was amplified using a pair of specific primers, including Ent1: 5'-TACTGACAAACCATTCATGATG-3' and Ent2: 5'-AACTTCGTCACCAACGCGAAC-3' (13).Enterococcus faecium EntfacYE Genome Analysis
The E. faecium EntfacYE genome was extracted using ethanol and propanol (precipitation method). Then, the extracted genome was wholely sequenced using Illumina Hiseq platform (Novogene, China) and de novo genome assembly as well as SPAdes algorithm. The bacterial genome was compre-hensively analyzed using RAST (rapid annotation using subsystem technology) online sequence analysis service (https://rast.nmpdr.org).
In general, the phenotypic assessment results verified the enterococcal isolate as E. faecium. To genotypically verify the isolates, the bacterial tuf gene was partially sequenced and the results were annotated in the DDBJ (DNA Data Bank of Japan) genomic database (accession Nos. LC580430 and LC580431) after primary analysis with BLAST (basic local alignment search tool) online service (https://blast.ncbi.nlm.nih.gov). Then, the bacterial whole genome sequence was analyzed, and the genome structure and subsystems were studied (Table 1). The bacterial whole-genome sequencing results were also annotated in DDBJ (BOPS 01000001–BOPS 01001574). EntfacYE genome subsystems included 23 various categories. In general, carbo-hydrates, amino acids, and protein metabolism cate-gories included the most-frequent and sulfur metab-olism, cell division and cell cycle, and phosphorus metabolism categories included the least-frequent subsystems in the EntfacYE genome. Antimicrobial resistance assessment results revealed resistance of E. faecium EntfacYE to vancomycin, erythromycin, clindamycin, cefoxitin, and ceftriaxone. Antibiotic resistance genes were divided into two major groups of genes with and without subsystems. Relatively, 49 antibiotic resistance genes were included in specific subsystems, while ten genes lacked specific subsy-stems (Tables 2 and 3). Furthermore, cadmium, cobalt, copper, zinc, and mercury resistance genes were identified.
Table 1. General information about E. faecium EntfacYE genome sequencing analysis
| Genome Annotation/Feature | E. faecium EntfacYE |
| DDBJ accession nos. | BOPS01000001–BOPS01001574 |
| Isolation source | Patient blood sample |
| Size (bp) | 3,624,552 |
| GC content (%) | 39 |
| Contig | 1 |
| Subsystems | 242 |
| CDS | 3957 |
| RNAs | 71 |
Table 2. Antimicrobial-resistance subsystems in E. faecium EntfacYE genome analysis
| No. | Subsystem | Feature |
|---|---|---|
| 1 | Copper homeostasis | Negative transcriptional regulator-copper transport operon |
| 7 | Copper homeostasis | Copper-translocating P-type ATPase |
| 1 | Copper homeostasis | Copper chaperone |
| 1 | Copper homeostasis | Copper tolerance protein |
| 2 | Bile hydrolysis | Choloylglycine hydrolase |
| 1 | Cobalt-zinc-cadmium resistance | Cobalt-zinc-cadmium resistance protein |
| 1 | Cobalt-zinc-cadmium resistance | Probable cadmium-transporting ATPase |
| 2 | Cobalt-zinc-cadmium resistance | Transcriptional regulator, MerR family |
| 2 | Mercuric reductase | PF00070 family, FAD-dependent NAD(P)-disulfide oxidoreductase |
| 2 | Mercuric reductase | Mercuric ion reductase |
| 2 | Mercury resistance operon | Mercuric ion reductase |
| 1 | Vancomycin tolerance locus | Sensor histidine kinase VncS |
| 2 | Vancomycin tolerance locus | ABC transporter, ATP-binding protein Vex2 |
| 1 | Vancomycin tolerance locus | Two-component response regulator VncR |
| 1 | Vancomycin tolerance locus | ABC transporter membrane-spanning permease, Pep export, Vex1 |
| 1 | Vancomycin Tolerance Locus | ABC transporter membrane-spanning permease, Pep export, Vex3 |
| 1 | Resistance to fluoroquinolones | DNA gyrase subunit B |
| 1 | Resistance to fluoroquinolones | DNA gyrase subunit A |
| 1 | Copper homeostasis: copper tolerance | Cytoplasmic copper homeostasis protein CutC |
| 1 | Fosfomycin resistance | Fosfomycin resistance protein FosX |
| 1 | Beta-lactamase | Metal-dependent hydrolases of the beta-lactamase superfamily I |
| 1 | Cadmium resistance | Cadmium efflux system accessory protein |
| 1 | Multidrug resistance efflux pumps | Multidrug resistance efflux pump PmrA |
| 1 | Multidrug resistance efflux pumps | Multiple antimicrobial extrusion protein (Na(+)/drug antiporter), MATE family of MDR efflux pumps |
| 1 | Mycobacterium virulence operon involved in protein synthesis (SSU ribosomal proteins) | SSU ribosomal protein S7p |
| 1 | Mycobacterium virulence operon involved in protein synthesis (SSU ribosomal proteins) | Translation elongation factor G |
| 2 | Mycobacterium virulence operon involved in protein synthesis (SSU ribosomal proteins) | Translation elongation factor Tu |
| 1 | Mycobacterium virulence operon involved in protein synthesis (SSU ribosomal proteins) | SSU ribosomal protein S12p |
| 2 | Mycobacterium virulence operon involved in DNA transcription | DNA-directed RNA polymerase beta subunit |
| 3 | Mycobacterium virulence operon involved in DNA transcription | DNA-directed RNA polymerase beta subunit |
| 1 | Mycobacterium virulence operon involved in protein synthesis (LSU ribosomal proteins) | LSU ribosomal protein L35p |
| 1 | Mycobacterium virulence operon involved in protein synthesis (LSU ribosomal proteins) | Translation initiation factor 3 |
| 1 | Mycobacterium virulence operon involved in protein synthesis (LSU ribosomal proteins) | LSU ribosomal protein L20p |
Table 3. Non-subsystem antimicrobial resistance genes in E. faecium EntfacYE genome
| Type | Length (bp) | Subsystem | Function |
| CDS | 531 | Uncharacterized | Protein YacP, similar to C-terminal domain of ribosome protection-type Tc-resistance proteins |
| CDS | 372 | Uncharacterized | Glyoxalase/bleomycin resistance protein/dioxygenase superfamily protein |
| CDS | 321 | Uncharacterized | Small multidrug resistance family (SMR) protein |
| CDS | 1713 | Uncharacterized | Heterodimeric efflux ABC transporter, multidrug resistance => LmrC subunit of LmrCD |
| CDS | 1773 | Uncharacterized | Heterodimeric efflux ABC transporter, multidrug resistance => LmrD subunit of LmrCD |
| CDS | 1920 | Uncharacterized | Tetracycline resistance, ribosomal protection type => Tet(M) |
| CDS | 900 | Uncharacterized | Cobalt/zinc/cadmium resistance protein CzcD |
| CDS | 486 | Uncharacterized | Teicoplanin resistance protein VanZ |
| CDS | 609 | Uncharacterized | D-alanyl-D-alanine dipeptidase (EC 3.4.13.22) of vancomycin resistance => VanX |
| CDS | 969 | Uncharacterized | D-lactate dehydrogenase VanH, associated with vancomycin resistance |
In recent decades, the resistance of most pathogens such as enterococci to available antibiotics has broadly increased. Enterococci cause a variety of infections, including opportunistic infections in hospitalized patients. In fact, E. faecium is one of the most important causes of nosocomial infections. Based on the reports, the mortality rate due to enterococcal bacteremia is estimated as 15–35% (14). In the present study, 23% of the bacterial genome was covered by subsystems, including various categories (Figure 1). The highest proportion was linked to the carbohydrates and protein metabolism groups, similar to a recent study on E. faecium R.A73 genome by Jeni et al. (2020) (15). Results from the present study demonstrated that the E. faecium EntfacYE strain isolated from a clinical blood sample was resistant to vancomycin, erythromycin, clindamycin, cefoxitin, and ceftriaxone and included various antibiotic-resistance genes. In recent decades, enterococcal resistance to vancomycin has increased, causing serious problems in treating infections (16, 17). In 2015, O’Toole et al. reported significant increases in vancomycin-resistant E. faecium isolates from Royal Hobart Hospital of Australia. Their study presented a shifting epidemic-ology pattern of VREfm from sporadic to endemic. They also showed the utility of isolates from asymptomatic patients to link epidemiological gaps between the patients (18). Lytsy et al. compared whole-genome sequences of vancomycin-resistant enterococci in three suspected outbreaks in Sweden during 2013–2015 using PFGE and MLST. They concluded that WGS-ANI was an easy method for this purpose. It was a user-friendly method, compared to MLST and PFGE. They recommended using WGS-ANI instead of PFGE for epidemiological outbreak investigations (19).
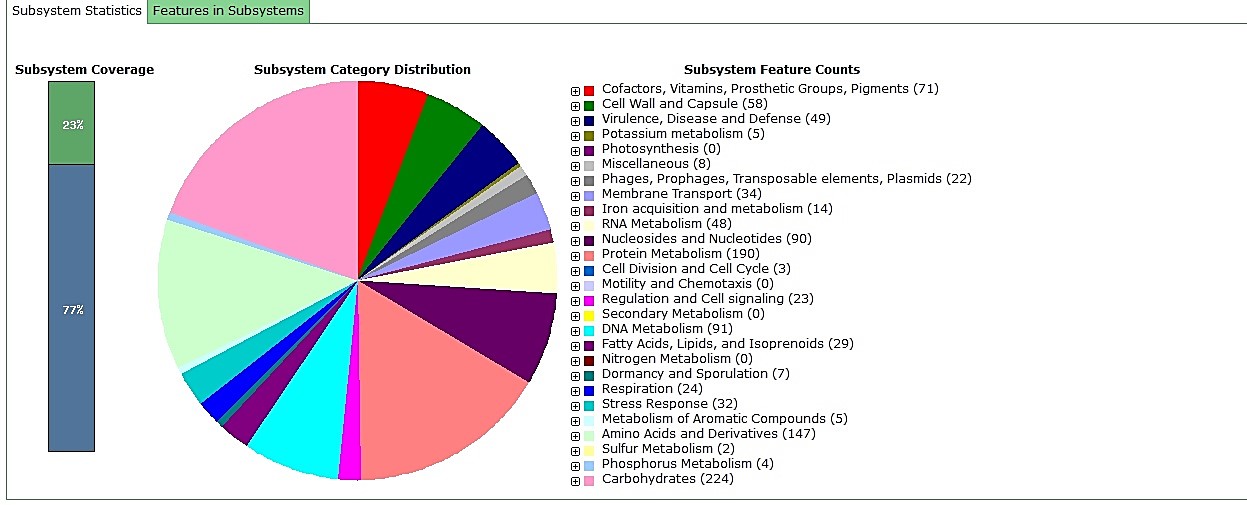
Figure 1. EntfacYE genome subsystem analysis using RAST
Based on the results from molecular studies, E. faecium is more resistant to routine antibiotics compared to E. faecalis, in which more than half of the enterococcal pathogens are resistant to common antibiotics such as vancomycin, ampicillin, and aminoglycosides (20). In this study, vancomycin resistance in EntfacYE isolate was due to the presence of van genes in the bacterial genome. A study in Iran showed that 12 out of 22 E. faecium isolates were resistant to vancomycin (21). In another study by Arbabi et al. (2016), more than half of E. faecium isolates were resistant to vancomycin (22). Active efflux pump is a common mechanism responsible for erythromycin resistance (23); thus, the presence of genes encoding efflux proteins in EntfacYE might lead to erythromycin resistance. In a study in Iran, 64% of enterococcal isolates, including E. faecalis and E. faecium, demonstrated resistance to erythromycin (24). Enterococci are intrinsically resistant to clinda-mycin; however, no relevant mechanisms have already been described (25). In this study, clindamycin resistance was associated with the ABC protein family. In a study in 2020, two genes involved in clindamycin resistance were identified in Bacillus licheniformis and Bacillus paralicheniformis (26). In the current study, resistance to cefoxitin and ceftriaxone was due to the presence of beta-lactamase genes. In a study by Rengaraj et al. (2016), 49% of S. aureus isolates were resistant to cefoxitin (27). In the present study, fosfomycin resistance genes were detected. This antibiotic is an alternative antibiotic against multi-drug-resistant bacteria (28). In a study on Pseudo-monas stutzeri, Soltani et al. (2015) reported that soils contaminated with heavy metals were potential sources for isolating metal-resistant strains (29). As heavy metals’ level increases in the environment due to modern industrial and agricultural activities, bacteria develop strategies to decrease intracellular concentrations of these toxic contaminants. There-fore, environments contaminated with heavy metals naturally contain microorganisms capable of resisting these metals (30). Plasmids mostly carry resistance-related genes and could spread resistance genes between bacteria through MGEs (e.g., the spread of plasmid genes from cadmium-resistant bacteria to other bacteria). In fact, heavy metal resistance is often associated with plasmids. However, this resistance is sometimes chromosomally originated (31). Copper homeostasis subsystem in E. faecium EntfacYE was reported in Salmonella enterica and Mycobacterium tuberculosis in 2019 (32).
Furthermore, bile salt hydrolysis determinants in E. faecium EntfacYE were investigated in Enterococcus strains isolated from artisanal dairy products in a study by Nami et al. (2019) (33). The presence of heavy metal resistance genes such as copper-zinc, cadmium, and mercury in the bacterial genome could be due to the gene transfer through plasmids or other MGEs. Accurate identification of Enterococcus species and their antibiotic resistance patterns is critical in providing effective treatments and appropriate medications (28, 34).
In this study, E. faecium EntfacYE genome analysis was carried out, focusing on antimicrobial resistance encoding genes. Further analysis of bacterial genomes, especially antibiotic resistance genes, can open new horizons for discovering novel medications other than common antibiotics and efficiently prevent the further spread of bacterial resistance.
The authors thank all the staff within the Microbiology Laboratory, School of Public Health, Tehran University of Medical Sciences for their help.
None.
Conflicts of Interest
The authors declared no conflict of interest.
Received: 2021/07/21 | Accepted: 2021/11/1 | ePublished: 2021/12/8
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
Copyright Policy
Iranian Journal of Medical Microbiology by Farname is licensed under CC BY-NC 4.0![]()
![]()
![]()
