




![]()
![]()
![]()
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
URL: http://ijmm.ir/article-1-1105-en.html



.
Coronaviruses in humans and animals are one of the causes of respiratory and intestinal infections (1). In November 2002، in Guangdong Province, southern China, A disease called SARS coronavirus spread that started with high fever and mild respiratory infection, but after a few days, symptoms of pneumonia developed. In addition, the disease is highly transmissible (2). In year 2012, the thirteenth of June, A 60-year-old Saudi man with Middle East respiratory syndrome coronavirus (MERS-CoV) symptoms was admitted to a private hospital in Jeddah. Now after many years, we are seeing the global outbreak of the Novel coronavirus again, the first reported of This new outbreak in December 2019 in Wuhan, China (3, 4). The outbreak spread rapidly and became pandemic, with evidence that the virus infected nearly 1.5 million people worldwide by 9 April 2020 (5). In this situation, the best way to deal with the disease is the fastest way. Nowadays, computer-assisted drug simulation methods can be performed on a computer, which is known as In-silico conditions. This method promises a future in the science of drug design (6). These methods have many advantages over traditional methods, including high speed and low cost. Using bioinformatics, we can test the performance of drugs on a computer, and bring the best of them into the laboratory. There are also drugs available for a variety of purposes, but it is possible that they have unknown capabilities that can be simulated by bioinformatics (7). Many recent studies begin with the question: Do different antiviral drugs, produced for different purposes, affect the coronavirus disease 2019 (COVID-19)? The answer to this question is very costly and time consuming using traditional methods (8-11). Molecular docking is one of the bioinformatics methods that can be performed under In-silico conditions. This method is able to check the orientation of the drug in the target and show the most appropriate condition (12). In this study, a number of antiviral drugs affecting RNA Polymerases were examined and their effect on SARS-CoV-2 RNA-dependent RNA polymerase was studied by docking. In addition, the optimal drug has been identified in terms of molecular docking, and its specific grid coordinates and interactions with the target have been investigated.
In this study, the input data includes drugs that have been approved and can be used against COVID-19 RNA polymerase (10). Also, similar structures that are related in terms of purpose and structure were examined. The online database used to search for drugs was the drugbank (13). 10 records were found and docking operations were performed by AutoDock Vina software (14). The SARS-CoV-2 RNA-dependent RNA polymerase structure (PDB ID: 7BTF) was saved from the PDB online database. Using AutoDockTools version 1.5.6 (ADT), polar and non-polar hydrogen were merged and Kollman charges were added (15, 16). In the next step, the structure was saved in PDBQT format. The following is the 3D structure of the 10 selected records, downloaded from drugbank online database. Structures were entered into the ADT software as a ligand, and were later automatically added Gasteiger charges, and was saved in PDBQT format. The docking operation was performed with two different size grids with coordinates center_x = 118.371, center_y = 125.314, center_z = 119.008, size_x = 106, size_y = 98, size_z = 126 and center_x = 144.957, center_y = 119.203, center_z = 120.929, size_x = 40, size_y = 40, size_z = 40. Discovery Studio 2020 Client software was used for the final docking analysis and the data were statistically analyzed (17). The results of molecular docking indicate an affinity for a position, which, given this value, can indicate a position that ligand can achieve with the least amount of energy. That's why we consider this value as the optimal binding energy. To control the results, molecular docking was repeated 10 times for Rifabutin. Also, molecular docking of Rifabutin on two similar SARS-CoV-2 RNA-dependent RNA polymerase structures (PDB ID: 7BZF, 7C2K) was performed, the first with Match Score of 45.51 and the second with Match Score of 10.45. The influenza C virus RNA-dependent RNA polymerase (PDB ID: 5D98) and hepatitis C virus NS5B RNA-dependent RNA polymerase (PDB ID: 1C2P) were used for negative control.

Table 1. Table of drug specifications and Affinity(kcal/mol) obtained from docking 1: Affinity A obtained from the grid with coordinates center_x = 118.371, center_y = 125.314, center_z = 119.008, size_x = 106, size_y = 98, size_z = 126 2: Affinity B obtained from the grid with coordinates center_x = 144.957, center_y = 119.203, center_z = 120.929, size_x = 40, size_y = 40, size_z = 40 (13).
| Name | Formula | ORGANISM | TARGET | Accession Number |
Binding Energy A1 )kcal/mol( |
Binding Energy B2 )kcal/mol( |
|---|---|---|---|---|---|---|
| Rifabutin | C46H62N4O11 | Escherichia coli | RNA-directed RNA polymerase | DB00615 | -9.2 | -7.0 |
| Sofosbuvir | C22H29FN3O9P | Hepatitis C Virus | RNA-dependent RNA-polymerase | DB08934 | -8.1 | -6.7 |
| Guanosine-5'-Triphosphate | C10H16N5O14P3 | Pseudomonas phage phi6 | RNA-directed RNA polymerase | DB04137 | -7.7 | -6.2 |
| Remdesivir | C27H35N6O8P | Zaire ebolavirus | RNA-directed RNA polymerase | DB14761 | -6.2 | -7.0 |
| C22H22F3NO5S | C22H22F3NO5S | Hepatitis C virus | RNA-dependent RNA-polymerase | DB07200 | -6.0 | -6.0 |
| Ribavirin | C8H12N4O5 | Influenza A virus & HPIV-2 | RNA-directed RNA polymerase | DB00811 | -6.0 | -5.7 |
| Taribavirin | C8H13N5O4 | Influenza A virus & HPIV-2 | RNA-directed RNA polymerase | DB06408 | -5.9 | -5.7 |
| Galidesivir | C11H15N5O3 | Zaire ebolavirus | RNA-directed RNA polymerase | DB11676 | -5.9 | -5.8 |
| C24H30N2O5S | C24H30N2O5S | Hepatitis C virus | RNA-dependent RNA-polymerase | DB07199 | -5.4 | -5.4 |
| Favipiravir | C5H4FN3O2 | Influenza A virus | RNA-directed RNA polymerase | DB12466 | -4.9 | -5.8 |
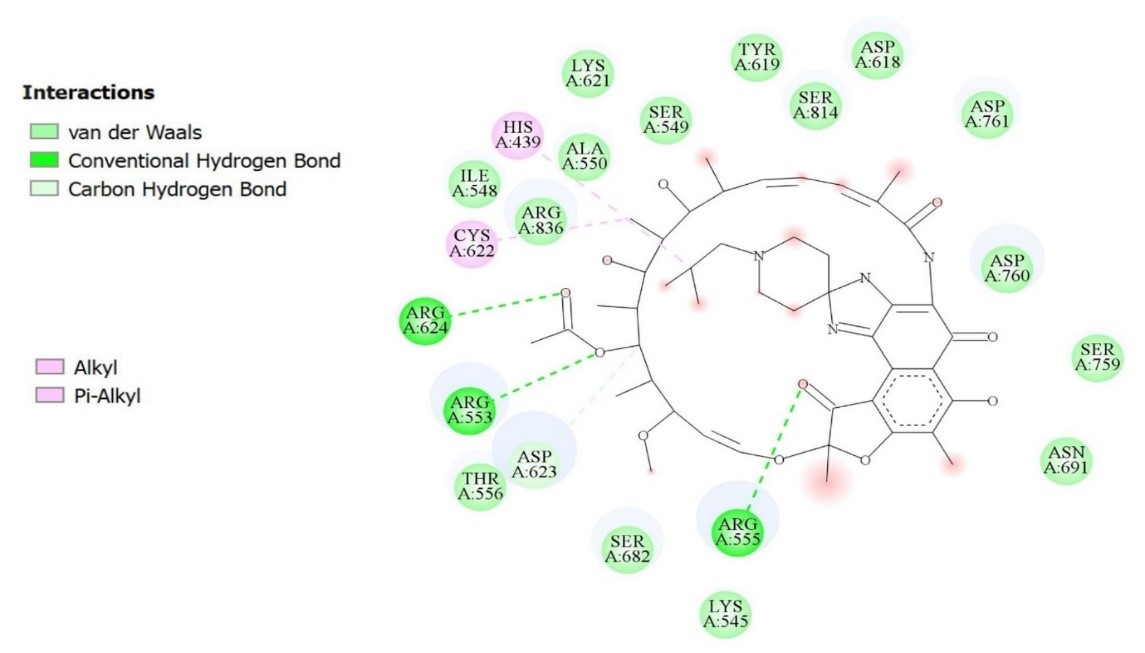
Figure 2. Interactions between the drug Rifabutin and the amino acids of the site of attachment to the target
In this study, during molecular docking, it was found that Rifabutin has the most negative affinity for SARS-CoV-2 RNA-dependent RNA polymerase binding, and also represents a new place for drug binding that could pave the way for the discovery of new drugs. molecular docking is one of the well-known methods in the exploration of new drugs. Finding the minimum amount of energy or optimization is an important technique in docking, according to which the ligand can find a position to connect with the least amount of energy. Docking also shows the best orientation of the ligand relative to the position of the protein binding site (18). The amount of affinity or more accurately the least optimal binding energy, obtained from the molecular docking of the drug Rifabutin is very significant because a recent study by docking targeting SARS-CoV-2 RNA-dependent RNA polymerase introduced Remdesivir with the affinity for the molecular docking of -7.803 kcal/mol as the best option, which shows this result. With a difference of -1.397 kcal/mol energies, the Rifabutin has a higher ability to bind (19). Also in another study, the molecular docking of Sophosbuvir as a ligand was performed by targeting SARS-CoV-2 RNA-dependent RNA polymerase, resulting in a molecular affinity of -9.3 kcal/mol Which is less than the amount obtained from the Rifabutin (10). It can also be deduced from molecular docking on control samples that the degree of similarity of the position to the target sample is directly related to the amount of binding energy. It is recommended that more study be done on the structure of this drug and that its binding site be studied.
As a result of this research in insilico conditions, it was found that the Rifabutin could be an inhibitor for SARS-CoV-2 RNA-dependent RNA polymerase, and that the drug-binding site had good conditions for the discovery of new drugs to inhibit SARS-CoV-2 RNA-dependent RNA polymerase of COVID-19.
In this regard, I would like to thank Dr. Payam Behzadi for his valuable comments.
Authors declared no conflict of interests.
- Cui J, Li F, Shi Z-L. Origin and evolution of pathogenic coronaviruses. Nature reviews Microbiology. 2019;17(3):181-92. [DOI:10.1038/s41579-018-0118-9] [PMID] [PMCID]
- Zhong N, Zheng B, Li Y, Poon L, Xie Z, Chan K, et al. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People's Republic of China, in February, 2003. The Lancet. 2003;362(9393):1353-8. [DOI:10.1016/S0140-6736(03)14630-2]
- York A. Novel coronavirus takes flight from bats? Nature Reviews Microbiology. 2020:1-. [DOI:10.1038/s41579-020-0336-9] [PMID] [PMCID]
- Zaki AM, Van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. New England Journal of Medicine. 2012;367(19):1814-20. [DOI:10.1056/NEJMoa1211721] [PMID]
- Coronavirus disease (COVID-2019) situation reports [Internet]. World Health Organization. 9 April 2020.
- Wadood A, Ahmed N, Shah L, Ahmad A, Hassan H, Shams S. In-silico drug design: An approach which revolutionarised the drug discovery process. OA drug design & delivery. 2013;1(1):3-7. [DOI:10.13172/2054-4057-1-1-1119]
- Meng X-Y, Zhang H-X, Mezei M, Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Current computer-aided drug design. 2011;7(2):146-57. [DOI:10.2174/157340911795677602] [PMID]
- Al-Tawfiq JA, Al-Homoud AH, Memish ZA. Remdesivir as a possible therapeutic option for the COVID-19. Travel Med Infect Dis. 2020;101615. [DOI:10.1016/j.tmaid.2020.101615] [PMID] [PMCID]
- Cai Q, Yang M, Liu D, Chen J, Shu D, Xia J, et al. Experimental treatment with favipiravir for COVID-19: an open-label control study. Engineering. 2020. [DOI:10.1016/j.eng.2020.03.007] [PMID] [PMCID]
- Elfiky AA. Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): A molecular docking study. Life Sciences. 2020:117592. [DOI:10.1016/j.lfs.2020.117592] [PMID] [PMCID]
- Elfiky AA. Anti-HCV, nucleotide inhibitors, repurposing against COVID-19. Life sciences. 2020:117477. [DOI:10.1016/j.lfs.2020.117477] [PMID] [PMCID]
- Morris GM, Lim-Wilby M. Molecular docking. Molecular modeling of proteins: Springer; 2008. p. 365-82. [DOI:10.1007/978-1-59745-177-2_19] [PMID]
- Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic acids research. 2018;46(D1):D1074-D82. [DOI:10.1093/nar/gkx1037] [PMID] [PMCID]
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of computational chemistry. 2010;31(2):455-61. [DOI:10.1002/jcc.21334] [PMID] [PMCID]
- PDB ID: 102L SARS-CoV-2 RNA-dependent RNA polymerase in complex with cofactors in reduced condition [Internet]. 8 April 2020.
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of computational chemistry. 2009;30(16):2785-91. [DOI:10.1002/jcc.21256] [PMID] [PMCID]
- Dassault Systèmes BIOVIA DS, 2020 Client, San Diego: Dassault Systèmes, 2020.
- Novikov FN, Chilov GG. Molecular docking: theoretical background, practical applications and perspectives. Mendeleev Communications. 2009;5(19):237-42. [DOI:10.1016/j.mencom.2009.09.001]
- Chang Y-C, Tung Y-A, Lee K-H, Chen T-F, Hsiao Y-C, Chang H-C, et al. Potential therapeutic agents for COVID-19 based on the analysis of protease and RNA polymerase docking. 2020. [DOI:10.20944/preprints202002.0242.v1]
Received: 2020/04/10 | Accepted: 2020/07/27 | ePublished: 2020/07/20
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
Copyright Policy
Iranian Journal of Medical Microbiology by Farname is licensed under CC BY-NC 4.0![]()
![]()
![]()
