




![]()
![]()
![]()
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
URL: http://ijmm.ir/article-1-2314-en.html



2- Department of Microbiology, Faculty of Medicine, Shahid Sadoughi University of Medical Sciences, Yazd, Iran
3- Gastroenterology and Hepatology Research Center, Institute of Basic and Clinical Physiology Sciences, Kerman University of Medical Sciences, Kerman, Iran
4- Faculty of Medicine, Kerman University of Medical Sciences, Kerman, Iran
5- Department of Internal Medicine, Shahid Sadoughi University of Medical Sciences, Yazd, Iran ,
Cancer stem cells (CSCs) have been emerged recently as a new concept in oncogenesis (1). It is identified that CSCs are rare cells, which are able to generate and develop tumors (2). On the other hand, CSCs differentiate to endothelial cells for angiogenesis in tumor cells, and facilitate the proliferation, colonization, and migration of tumor cells (3, 4). Oncoviruses, utilize the host cell effectors and signaling pathways that control the host cell condition for effective infection (5, 6). Also, some reports assessed the role of the host cell kinases as the main signaling factors in the viral replication and assembly (7, 8). They modulate variety of the cellular signaling pathways, which are related to the regulation of gene expression and homeostasis of the cell, containing mRNAs and proteins synthesis (9).
The c-Jun N-terminal kinases (JNKs) as members of the MAPK family are identified to control cell proliferation, migration/invasion, apoptosis, autophagy, necroptosis, pyroptosis, ferroptosis, and cell survival-mediated cancer therapeutic resistance (10). JNKs are a kind of stress-activated protein kinases (SAPKs), which can be triggered by radiation, growth factors, cell stress, and inflammatory cytokines (11). There are three genetic loci in human genome that encode JNK1-3 through alternative splicing of the related pre-mRNAs, each one has 2 to 4 isoforms (12). Three genes, namely JNK1 (MAPK8 or SAPK1), JNK2 (MAPK9 or SAPK2) , and JNK3 (MAPK10 or SAPK3), encode 10 different splice variants of JNK (13). JNK1, JNK2 and JNK3 are located on chromosomes 10q11.22, 5q35.3 and 4q21.3, respectively. Although JNK1 and 2 are generally expressed in all human tissues, JNK3 is highly expressed in the brain cells and lower expression levels is seen in the testis and heart cells (14). JNKs have a proved redundancy function in phosphorylation of their substrates that consist of c-Jun, ATF2, JunD, polycomb repressive complex 1 (PRC1) (15), Akt (16), FoxO4, c-Myc, p53, NFATc2, STATs (17), IRS-1, histone H3 (18), SIRT1 (19), and some other proteins (9).
Transforming growth factor‐β (TGF‐β) signaling pathway plays vital roles in the cellular biological processes such as apoptosis, proliferation, epithelial‐mesenchymal transition, differentiation, vascular disorders, organ fibrosis, and cancer (20). With regard to the tumorigenesis, it displays a dual function by exhibiting anti‐tumor activity through inducing apoptosis and suppressing cell cycle (21). Moreover, it also acts during the late stages of oncogenesis and functions as a tumor inducer by enhancing tumor invasiveness and metastasis (22). TGF‐β cytokines (TGF‐β1, TGF‐β2, and TGF‐β3) bind to the type I and II serine‐threonine kinase receptors and activate two diverse downstream pathways; SMAD‐dependent and SMAD‐independent. This signaling pathway can be disturbed by factors such as mutation and microbial interference (23).
Discrepancy in the TGF-β signaling might be implicated in the progression of tumors in the viral persistent infection. For example, in the development of HBV persistent infections, HBx oncoprotein changes TGF-β signal pathway from the TβRI/pSMAD3C tumor-inhibitor axis to the JNK/pSMAD3L tumorigenic path in the early phase of the persistent infections (24). Furthermore, the role of JNK1 over JNK2 in the pathogenesis of some human diseases such as diabetes is its activation by cytokines. It is a key mediator in the transition between obesity and type 2 diabetes (T2D). Its role in free fatty acids and hyperglycemia, lung fibrosis, and cancer has been revealed (25).
Recognition of the pathogen-associated molecular patterns (PAMPs) by the pattern recognition receptors (PRRs) stimulates innate immune cells and subsequently induces the chemokines and cytokines expression that directly target microbes. Amongst the PRRs, the Toll-like receptors (TLRs) have been well studied as the stimulation of all 10 TLRs activates the MAPK and NF-κB signaling pathways that are necessary for an effective immune system response against infections.
The JNK pathway plays a key role in oncoviruses replication. It can be triggered through viral infection and is involved in the replication of some viruses including herpes viruses (26) and rotaviruses (27). However, it is revealed that JNK activation can induce (28) or suppress (29) the viral replication and represent a critical factor in the cell death triggered via certain chemotherapeutic compounds (30) and stress responses (31, 32). Therefore, it has the potential to either induce or suppress the viral replication (33). In this review we aimed to study and underscore the effects of oncoviruses including HBV,HCV, HTLV,HPV, KSHV, and EBV on JNK signaling pathway (34).
1-1- Hepatitis B virus
Hepatitis B virus (HBV) is a hepatotropic oncovirus containing a partially double-stranded circular DNA genome, which can be integrated into the host genomic DNA and causes hepatocellular carcinoma (HCC). This infection is one of the main factors in developing HCC; as epidemiological studies have revealed that around 50% of all HCC cases, are related to HBV infection (35).
HBx is a non-structural protein encoded by HBV genome. It is involved in the induction of several host cell signaling pathways, such as the RAF/RAS/MAPK (36), JAK signal transducer and activator of transcription (STAT) (37) (38), mitogen-activated protein kinase 1 (MEKK1)/JNK (39), phosphoinositide 3-kinase (PI3K)/AKT (40), and Notch1 signaling (41) pathways, leading to the development of tumor and cell survival.
Autophagy is a protein degradative mechanism, which is essential for both the normal cellular hemostasis and eliminating the intracellular invading pathogens (42). Interestingly, several viruses including HBV are capable of regulating the cell autophagy pathway. HBx triggers autophagosome formation of the class I phosphatidylinositol 3-kinase (PI3K)/AKT/mTOR signaling pathway (43). In turn, the class III PI3K (VPS34)/beclin-1 signaling have shown to be essential for the HBx-related autophagosome development. Additional studies have demonstrated that HBx does not have any effect on the beclin-1 and VPS34 expression but it suppresses the coactivity of beclin-1/Bcl-2 through the JNK signaling (44). Furthermore, transfection studies revealed that HBx induces production of reactive oxygen species (ROS) (39). Moreover, suppression of ROS activity by the ROS scavenger N-acetylcysteine (NAC) inhibits both autophagosome development and JNK function (43).
ROS-JNK signaling also demonstrated to play a key role in HBV-triggered autophagy and successive HBV replication (43).
Interestingly, the increase in the TGF-β level might be associated with the progress of HCC in the persistent HBV infection (45). In the development of HBx-induced carcinogenesis, HBx causes shifting of TGF-β signaling from the pSMAD3C/TβRI tumor-inhibitor pathway to the pSMAD3L/JNK tumor-inducer pathway in the early chronic hepatitis B (CHB) infection (46). Based on the effects of HBx on TGF-β, some studies showed that suppression of HBx-related activation of pSMAD3L/JNK can make HCC cells sensitive to TGF-β and enhances its anti-tumor activity (45).
Akt signaling has been revealed to be involved in the modulation of hepatic glucose metabolism (47). HBx can affect the Akt phosphorylation by the JNK signaling, resulting in induction of stress and inflammation in the hepatocytes (48). The JNK is recognized to mediate serine phosphorylation of insulin receptor substrate 1 (IRS-1), therefore, decreases the function of IRS-1 and its downstream signaling pathway including Akt (49). Also, a report has revealed that HBx-overexpressing in the liver of transgenic mice (HBxTg) can reduce the Akt phosphorylation and upregulate the JNK1 phosphorylation, while JNK1 phosphorylation was reduced through null mutation of Inducible Nitric Oxide Synthase (INOS) (50, 51). Moreover, previous in vitro study has shown that HBx induces the cell death by triggering the JNKs- MLK3-(mitogen-activated protein kinase kinase 7) MKK7 signaling that induces FasL expression (48). Another study showed that HBx can suppress the cell proliferation and increase the apoptosis by Fas/FasL modulation in the rat epithelial cells (52). Interestingly, somez studies have reported that HBV particles, HBsAg, and HBeAg are able to interfere with the TLR signaling that can affect the JNK activation (53).
Another HBV-encoded protein, HBsAg, can influence the TLR4 signaling pathway. Studies have shown that the LPSinduced phosphorylation of p38 and JNK, and the degradation of IkB-α can be suppressed by HBsAg. Also, it has been reported that LPS-induced production of cytokines such as IL-6, IL-12, and TNF-α could be suppressed via HBsAg (54).
HBsAg may suppress the IL-12 production, especially via decreasing the effect of the MAPK-JNK pathway. This theory was investigated in monocytes/macrophages of the chronic hepatitis B patients and HBsAg-negative control healthy group (55). The inhibited TLR2-mediated JNK activation and IL-12 secretion were also reported in the CHB patients (55). The mechanisms that HBsAg inhibits the JNK–MAPK signaling is not well known yet, but suggested to be via directly affecting the JNK or its upstream effectors or JNK function modification by influencing the inhibitory pathways in the cells (56). To explain the mechanisms in detail more assessment is needed.
It was reported that Adaptor Related Protein Complex 1 Subunit Mu 1 (AP1M1) is upregulated in the HBV‑transfected HepG2.215 cell line and AP1M1 silencing in HepG2.215 cells causes the suppression of cell proliferation. Data proposed that AP1M1 is a crucial factor related to the progression of the liver cancer related to the HBV infection. Furthermore, it was reported that HBx increased the AP1M1 expression in a JNK‑dependent manner. The AP1M1 increased protein kinase B phosphorylation, resulting in the acceleration of the cell proliferation (57). Major biological pathways involved in transforming activities of HBV are shown in Figure 1.
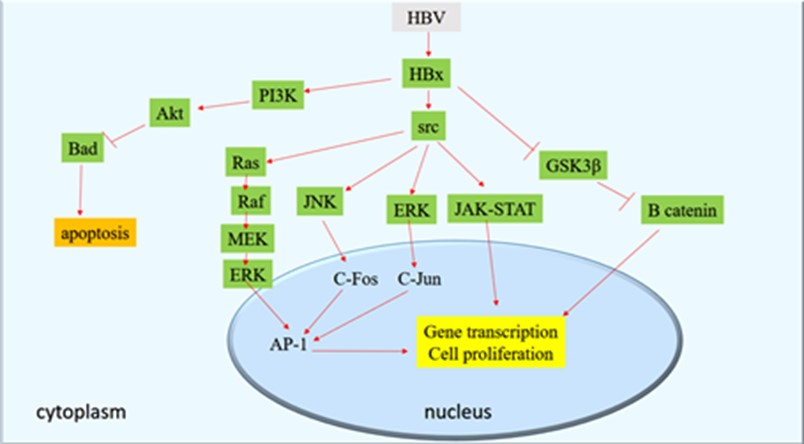
Figure 1. Major biological pathways involved in transforming activities of HBV (Designed by Authors, 2024)
1-2- Hepatitis C virus
Hepatitis C virus (HCV) is a single-stranded RNA virus that cannot integrate into the host cell genome. Since the virus life cycle is in the cytoplasm, its oncogenesis is likely to be limited in the cytoplasmic compartments, as indirect mechanisms (58). The immune system response to the kill the infected hepatocytes and following the regeneration process as well as chronic inflammation is crucial for the development and progression of the HCC related to HCV. In transgenic mice it has been demonstrated that, the HCV core protein can be implicated in the HCC development and might play a key role in HCC progression (59).
The HCV core protein has the ability to upregulate the JNK pathway, leading to the stimulation of the hepatocyte regeneration through decreasing p21 levels and increasing c-Myc expression (60). Moreover, the HCV core protein has the ability to upregulate the JNK pathway through a regulatory process involving vascular endothelial growth factor (VEGF) (61). The inflammatory response stimulated via HCV infection leads to the tumor progression, although it is a part of the host immune system. Commonly, HCV disrupts the host immune system and causes chronic inflammation in association with the persistent infection (62). Chronic inflammation leads to an abnormal frequent reaction and triggers the liver cell apoptosis and proliferation that is related to the subsequent progression of HCC (63, 64). Reduction of p21 half-life and high expression of c-Myc was reported in HCV core transfect cell culture as well as HCV core transgenic mice (65).
Moreover, another encoded HCV protein, NS5A, appears to act as positive modulator of the JNK signaling through interacting with TNF receptor associated factor (TRAF) that might be involved in the HCV pathogenesis (66). In an experiment, Lin et al., revealed that HCV increases the secretion of TGF-β from hepatocytes in the overexpressed ROS and JNK condition (67). Binding of TNF-α to TNF receptor 1, forms complex I that involves TNFR death domain, TRAF2, cIAP-1, cIAP2, RIP1 and a dimeric ubiquitin binding enzyme combined of Ubc13. In turn, cIAP-induced K63 ubiquitination of RIP1 activates TGF-β activated kinase 1 (TAK1) that triggers JNK by MAP kinase kinase 4/7 (MKK4/7) (68, 69). Additionally, this complex participates to the ROS signaling by decreased Rac1 and nicotinamide adenine dinucleotide phosphate oxidase.
The increased level of ROS results in the extended stimulation of JNK through restricting the JNK phosphatases (70). Other studies have revealed the correlation between the TGF-β-associated Smad and JNK pathways (71) that can inhibit TGF-β-related growth suppression, in a manner required for oncogenesis (72). In the cell nucleus, JNK1 can modulate nuclear function of the Smad complexes that enhances binding of the Smad complex to DNA and mediates various gene responses to TGF-β to modulate the ECM-related genes expression that is involved in oncogenesis (73). Also, TGF-β stimulates indirect or non-Smad signaling pathway of TAK1 and JNK (74) such as regulation of transcriptional responses, and phosphorylation of cytoplasmic Smad proteins linker that is crucial in association of TGF-β signaling with the JNK pathway (75). Major biological pathways involved in transforming activities of HCV are shown in Figure 2.
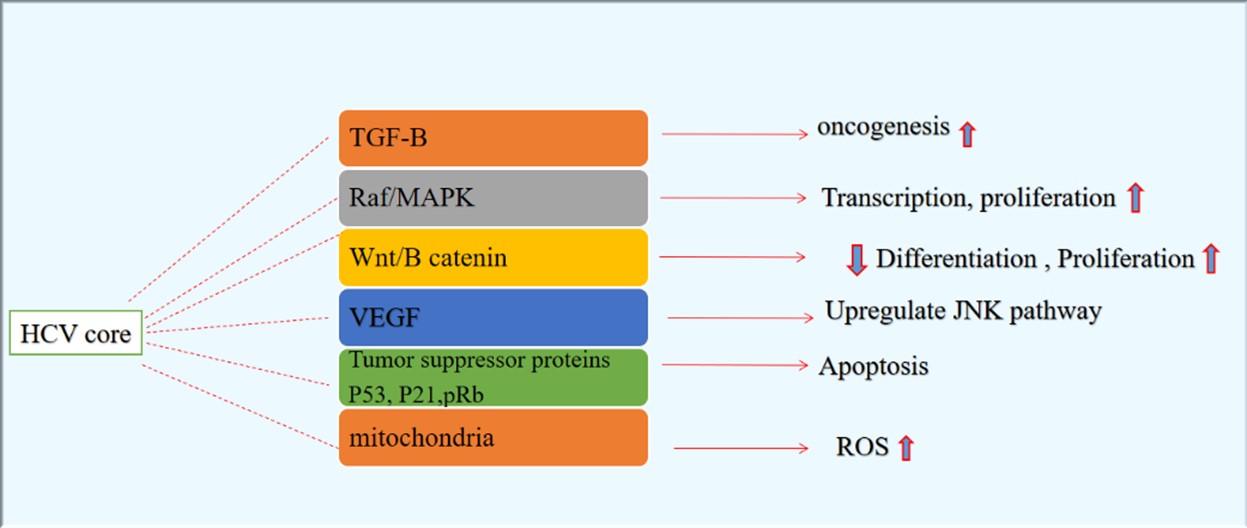
Figure 2. Major biological pathways involved in transforming activities of HCV (Designed by Authors, 2024)
1-3- Human T cell Leukemia Virus-1
Adult T-cell leukemia (ATL) has been recognized as a T-cell malignancy that is related to the human T-cell leukemia virus type 1 (HTLV-1) chronic infection (76). Matrix metalloproteinases (MMPs) degrade extracellular matrix that is a critical mechanism in the invasiveness and metastasis of cancers (77). The MMP-7 also known as matrilysin, is a “minimal domain MMP” with ubiquitin-like activity towards the extracellular matrix components. Previous studies have revealed the high expression of MMP-7 in the ATL cells and lymph nodes of HTLV-1-infected patients as well as T-cell lines (78). It has also been reported that MMP-7 expression can be upregulated following the HTLV-1 infection thorough the HTLV-1 Tax protein, resulting in migration of the HTLV-1infected T cells (79). The Tax protein, as a transactivator of the cellular genes, can upregulate MMP-7 promoter activity but MMP-7 promoter activity can be down-regulated via deletion of the activator protein-1 (AP-1) binding site (80). It has been reported that Tax can bind to the AP-1 binding site of MMP-7 similar to JunD. Inline, studies showed high levels of AP-1 binding proteins, such as JunD in T-cell lines associated with ATL (related to HTLV-1) cells (81). The MMP-7 activation might be decreased through the inhibition of JunD signaling pathway or be increased via JunD homodimers (82). Previous studies have shown that blockade in JunD through the small interfering RNA (SiRNA) can inhibit MMP-7 expression in the HTLV-1-infected T-cell lines (83). Overall, these studies suggest that the stimulation of MMP-7 through Tax can be modulated via JunD and also MMP-7 can promote visceral invasion in the ATL patients.
Another protein encoded by HTLV-1 is bZIP (HBZ) that is suggested to be associated with proliferation of T lymphocyte. The C-terminal zipper domain of HBZ interacts with c-Jun (84) resulting in a decrease in c-Jun DNA-binding activity and inhibition of transactivating activity of AP-1. It is not clear yet that the inhibitory effect of HBZ-SP1 is due to its weak DNA-binding activity or its ability to interact with other cellular elements to inactive nuclear bodies (85). A HBZ-SP1 mutant with specific residues in the regulatory and DNA-binding domain, which was replaced for conforming c-Fos amino acids, increases the potential activity of the DNA-binding of the HBZ-SP1/c-Jun heterodimer (86). Moreover, this study revealed that the inhibition of c-Jun activity in vivo is mostly related to the sequestration of the HBZ-SP1-c-Jun to HBZ-NBs.
The N-terminal motif (LXXLL) of HBZ mediates the activation of TGF-β signaling pathway. The HBZ increases the p300/Smad3 complex remarkably and overcomes the suppression of the TGF-β response by Tax protein. Similar to TGF-β, HBZ expression increases the transcription of Pdgfb, Ctgf, Foxp3, Sox4, Tsc22d1 and Runx1 genes but suppresses the transcription of Id2 gene. In naive T cells, HBZ induces the Foxp3 expression by the Smad3-dependent TGF-β signaling pathway. Data were proposed that HBZ enables HTLV-1 virus to convert the infected T lymphocyte cells into the regulatory cells by enhancing the TGF-β signaling and Foxp3 gene expression that might help HTLV-1 persistency. Major biological pathways involved in transforming activities of HTLV are shown in Figure 3.
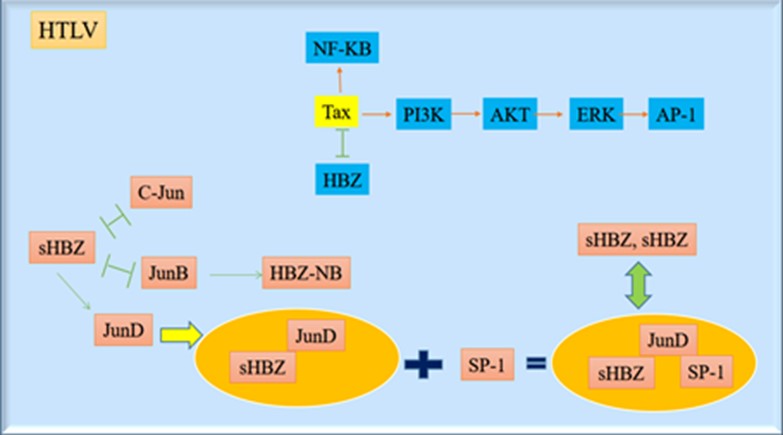
Figure 3. Major biological pathways involved in transforming activities of HTLV (Designed by Authors, 2024)
1-4- Epstein-Barr virus
Epstein-Barr virus (EBV) belongs to the Herpesviridae family and is related to several human malignancies such as nasopharyngeal carcinoma (NPC), Burkitt’s lymphoma (BL), and gastric carcinoma (87). After primary infection, EBV can appear as a latent infection for a lifelong in B cells with potential reactivation in the specific situations, especially immune suppression (88). Switching EBV infection from the latency phase to the lytic cycle is mediated via the expression of Zta immediate-early protein, which is also named as ZEBRA and BZLF1 (89). The Zta protein acts as a transcription activator that has partially the same amino acid sequence as leucine zipper family of transcription elements such as c-Fos and c-Jun (37).
EBV encodes latent membrane protein 1 (LMP1), that is an oncoprotein considerably expressed in BL and NPC (90). The LMP1 has been recognized as an integral membrane protein that is crucial for the EBV-induced B-cells and fibroblasts immortalization and transformation (91). Studies using LMP1-expersion cell line have revealed that LMP1 increased ROS through Akt/PI3K signaling pathway resulting in increased expression of the genes involved in glycolysis (92). A study revealed that LMP1 may activate NAD(P)H oxidases (NOX) and upregulate ROS in EBV-associated malignancies (93). Assessment of NOX expression revealed that NOX modulatory subunit p22phox expression increased pronouncedly in the cell transected by LMP1 through JNK-dependent and -independent pathways (94).
The LMP1 triggers the activation of the AP-1 transcription factor, a heterodimer of Jun/Fos or homodimer of Jun/Jun proteins. The effect of LMP-1 on AP-1 is through the JNK pathway, except for the extracellular signal-regulated kinase (Erk) signaling pathway (95). Therefore, LMP1 can induce the activation of the c-Jun N-terminal transactivation domain that is identified to be triggered through the JNK-mediated phosphorylation (96).
Previous experiments demonstrated that C-terminal domain of LMP1 is crucial for the activation of AP-1. Moreover, the study using LMP1 mutant showed that JNK-induced transactivation of AP-1 is the direct result of LMP1-stimulated signaling (97). Additionally, through a tetracycline-modulated LMP1 allele, researchers showed that JNK is an element of the LMP1 signaling pathway in B cells immortalization (98). Taken together these data demonstrated a new function of LMP1 in the JNK/SEK/AP-1/c-Jun pathway that increases our knowledge regarding the potential function of LMP1 in immortalization of B cells (99). Major biological pathways involved in transforming activities of EBV are shown in Figure 4.
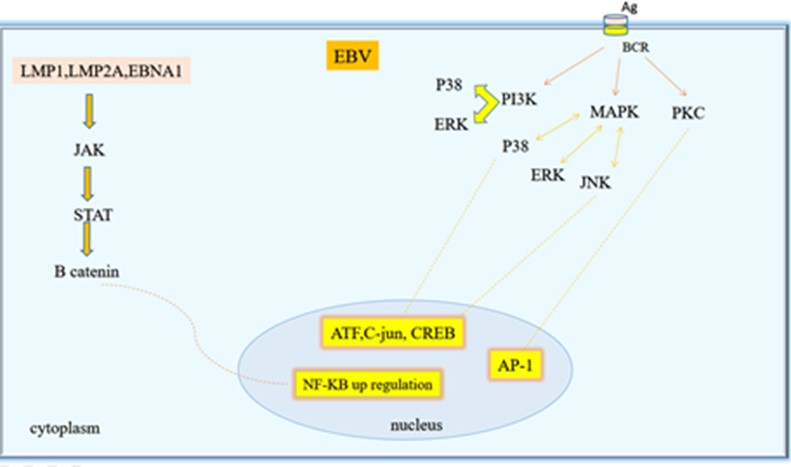
Figure 4. Major biological pathways involved in transforming activities of EBV (Designed by Authors, 2024)
1-5- Human papillomaviruses
Human papillomavirus (HPV) infection is a major cause of benign and cancerous proliferative lesions of the mucosal epithelium. In 2018, cervical cancer was known as the second most common cancer in women living in developing countries with approximately 311000 death (100). The epidemiological and experimental studies demonstrated that HPV infection is the main factor in cervical cancer development and progression (101). Especially, the HPV types 16 and 18 have been known as the most high-risk HPVs and are the main contributing agents in more than 90% of all cervical cancers.
Dysregulated expression of HPV oncoproteins E6 and E7 is the most important etiology of cervical cancer (102). Furthermore, stimulation of the Wingless type (Wnt)/β-catenin signaling pathway has been suggested to be involved in cervical tumorigenesis following the triggered signal in a multi-step mechanism (103), although the details of molecular process of cervical cancer development remain unclear. Wnt-11 has been known as an enhancer of the Wnt signaling pathway that plays a critical role in cervical oncogenesis (104). Wnt-11 and HPV E6 protein have been reported to be upregulated in a condition, which is related to the development of cervical cancer and is significantly associated with cancer phase, metastasis, tumor measures, and HPV infection (105). Wnt-11 expression reported to be significantly correlated with HPV E6 expression in most of the cervical cancer samples (106). Also, Wnt-11 reported to be accompanied with the expression of P-JNK1, cell propagation, developed cervical cancer, and invasion (107). These outcomes suggest that the upregulated expression of Wnt-11 reported in tumor cells can result in the phosphorylation and stimulation of JNK-1 and potentially enhances the cervical cancer cell propagation and invasion by triggering the JNK/Wnt signaling (108). Therefore, Wnt-11 can be a novel target for the treatment of cervical cancer.
The E6 and E7 oncoproteins of HPV-16 promoted the activation of Akt, P85S6K, P70S6K, mTOR, JNK, and c-Jun. Knockdown of C-Jun by the specific siRNA, eliminated the HPV-16 oncoprotein-induced HIF-1α, VEGF, and IL-8 expression. Additionally, HIF-1α protein stability was promoted by HPV-16 oncoproteins via blocking the proteasome degradation pathway, but knockdown of c-Jun abolished this effect (109). Furthermore, E6 and E7 oncoproteins of HPV-16 amplified the quantity of c-Jun binding to HIF-1α protein.
Levan et al., confirmed that the cellular protein NFX1-123 is a regulator of the epithelial cell differentiation and consequently provides insight into the association between the HPV life cycle and epithelial differentiation (110). They recognized that NFX1-123 specifically affected the JNK signaling pathway and vigorously activated proteins within that cascade (111). Major biological pathways involved in transforming activities of HPV are shown in Figure 5.
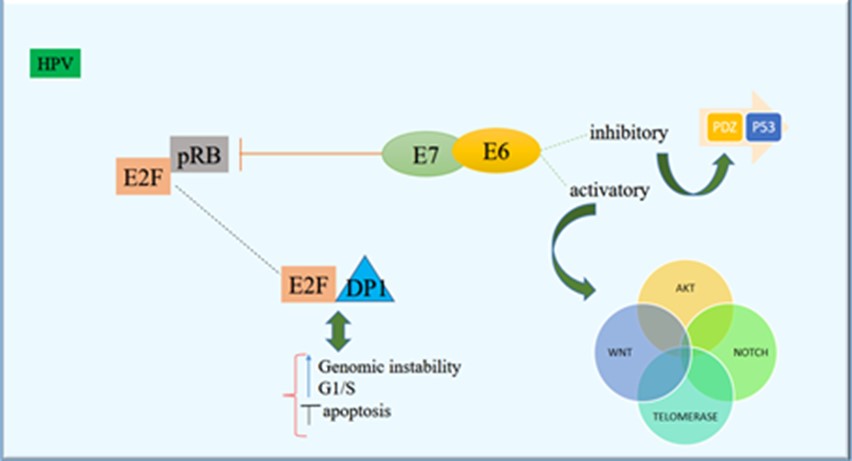
Figure 5. Major biological pathways involved in transforming activities of high-risk HPVs (Designed by Authors, 2024)
1-6- Kaposi’s sarcoma-related herpesvirus
Kaposi’s sarcoma-related herpesvirus (KSHV) or human herpes virus type 8 is identified to be associated with Kaposi’s sarcoma, a cancer of endothelial cells that is commonly seen in AIDS patients (112). In recent studies it has been demonstrated that KSHV can activate the JNK/ERK/MEK and p38 MAPK signaling through the primary infection of human umbilical vein endothelial cells known as HUVEC, and these pathways control the viral entry processes and viral lytic infection (113). The KSHV proteins can suppress the autophagy at various phases in different cells that clearly reveals to be associated with v-BCL2, FLIP/v-CFLAR and K7 (114, 115).
It has been reported that v-BCL2 and v-CFLAR/FLIP inhibit autophagy in the early phase by suppression of autophagosome maturation. Subsequently, it can inhibit differentiation of monocytes, which is reported in KSHV infection (116). Moreover, FLIP/v-CFLAR signaling can act similar to the cellular FLIP/CFLAR that might suppress the JNK/MAPK activity by interacting with the MAP2K7/MKK7 (117).
Noticeably, it has been shown that a deletion in the coding region of JNK1/MAPK8 or JNK2/MAPK9 genes may result in the equilibrium of FLIP/CFLAR. Moreover, JNK/MAPK suppression could be an approach for stabilizing the CFLAR viral protein (12, 118). Furthermore, FLIP/v-CFLAR can trigger NF-ĸB and a reverse association has been reported between these two signaling pathways (119).
v-BCL2, as its cellular homolog, suppresses the autophagy through binding to BECLIN1 but independently from BCL2. v-BCL2 may not be apart from BECLIN1 in the starvation situation. Certainly, v-BCL2 does not have any site of phosphorylation via JNK1/MAPK8 (120). Although, cellular BCL2 phosphorylation is facilitated through the JNK1/MAPK8, data showed that KSHV infection basically decreases the function of the isoform of JNK2 p54/MAPK9 (121). It is important to clear that v-BCL2 may interact with kinases that modulate JNK/MAPK phosphorylation and associate with the autophagy suppression via decreasing the ATG5 and CAST. Major biological pathways involved in transforming activities
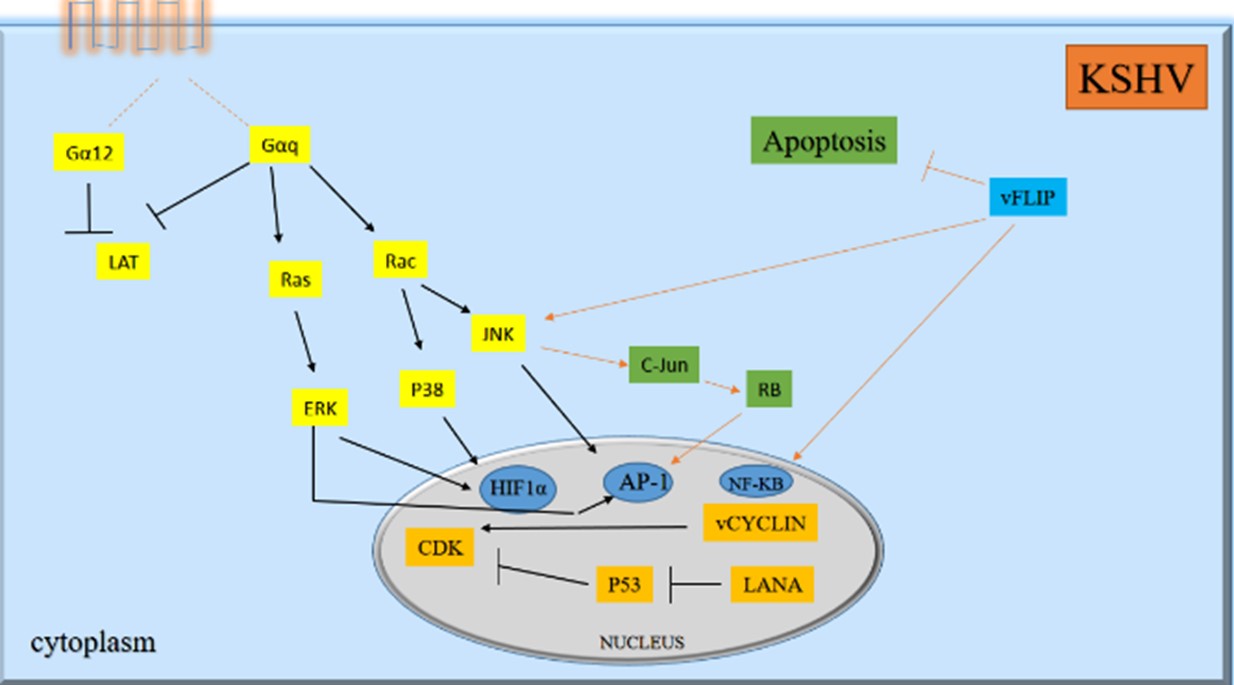
Figure 6. Major biological pathways involved in transforming activities of KSHV (Designed by Authors, 2024).
The JNK pathway plays a key role in the oncogenesis mechanism of oncoviruses by influencing both oncogenic events and tumor suppressive mechanisms. It has been shown that HBx/HBs Ag of HBV, LMP-1of EBV, v-BCL2 and v-CFLAR of KSHV, NS5A of HCV, HBZ of HTLV, and E6/E7 of HPV 16 may affect the function of JNK signaling pathway. Determining the viral proteins that affect the JNK signaling pathway may be a key factor to understand the role of the viral proteins during the carcinogenesis by this mechanism and may provide even greater insight into the cancers related to these viruses. The JNK signaling pathway will thereby add to the other targets available for the malignancy treatment related to the oncoviruses. Further studies are needed to clarify the impact of JNK signaling pathway in the viruses-related cancers.
Not applicate.
Ethical Approvel
Not applicate.
Authors’ Contributions
E.B. and Y.G.: concept, study design, supervision and writing-original draft preparation. J.C. and M.N.: writing-original draft preparation. A.K.: writing-review and editing. The author(s) read and approved the final manuscript.
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflicts of Interest
No conflicts of interest were declared by the authors.
Received: 2023/12/11 | Accepted: 2024/01/29 | ePublished: 2024/08/18
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
Copyright Policy
Iranian Journal of Medical Microbiology by Farname is licensed under CC BY-NC 4.0![]()
![]()
![]()
