




![]()
![]()
![]()
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
URL: http://ijmm.ir/article-1-2263-en.html



2- College of Health Sciences, University of Human Development, Al Sulaymaniyah, Iraq ,
3- Department of Infection Biology & Microbiomes, Faculty of Health & Life Sciences, University of Liverpool, The United Kingdom
The large family of RNA viruses known as Coronaviridae is responsible for various diseases, from the common cold to some severe ones like Severe Acute Respiratory Syndrome (SARS-CoV) and Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2), which produces the disease named coronavirus disease-2019 (COVID-19).
The Coronavirus has a 26-32 kb single-stranded positive-sense RNA. Based on phylogenetic analysis and the International Committee of Taxonomy of Viruses (ICTV) 2023 reports, the Coronaviridae family has three subfamilies (Letovirinae, Orthocoronavirinae, and Pironaviruses), which are classified into six genera. Alphacoronavirus (αCoV), Betacoronavirus (βCoV), Gammacoronavirus (γCoV), and Deltacoronavirus (δCoV) are into Orthocoronavirinae subfamily and Alphapironavirus belong to pironaviruses while Letovirinae includes the Alphaletovirus genus. Coronaviruses include more than 30 species that infect humans, mammals, birds, and fish. However, Orthocoronavirinae can infect a wide range of birds and mammals.
Both humans and animals are infected with Coronaviruses (αCoV and βCoV). Thus 1960, the first human coronavirus was discovered (1, 2). In humans, 229E, NL63, OC43, HKU1, MERS, SARS-CoV, and SARS-CoV-2 are the eighth reported coronaviruses (3). Demonstrated that SARS-COV was caused an outbreak of lineage 2B βCoV, a virus with bat origin in 2002–2003, while coronavirus-related Middle East Respiratory Syndrome (MERS) was caused by a class C beta-coronavirus with camel origin in 2012 (4).
Due to its respiratory symptoms, a pneumonia case in Wuhan, China, that was initially reported in late December 2019 and later determined to be caused by a coronavirus was labeled as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) by the World Health Organization (WHO) (5, 6). It has been established that the new coronavirus for the 2019 outbreak is a hybrid virus from the bat coronavirus and other animals (7). SARS-CoV-2 shares 82%, 89%, and 96.3% nucleotide similarity with SARS-CoV, SARS-like CoV ZXC21, and bat coronavirus RaTG13, respectively, which confirms its zoonotic origin (8, 9).
SARS-CoV-2 RNA-dependent RNA polymerase (RdRp) complex is used to replicate the virus genome and transcribe its genes, so it serves as a critical component in the replication/transcription machinery. RdRp complex is a pivotal target for antiviral drugs such as Remdesivir, Favipiravir, Galidesivir, and Ribavirin. Mutations in RdRp can potentially modify the protein's structural stability, influencing its interactions with RNA patterns or other transcription/replication machinery elements. Consequently, these alterations may impact the mutation rate. Thus, these alterations may affect the mutation rate (10).
The SARS-CoV-2 nucleocapsid (N) protein is crucial in the coronavirus life cycle, engaging in essential activities post-virus invasion. Its significance has garnered considerable attention in vaccine and drug development (11). The envelope (E) protein, despite being the smallest structural protein, remains enigmatic. It can form as an intracellular ion channel or viroporin, functioning as a standalone virulence factor that could lead to host cell death and trigger a cytokine storm. This characteristic positions E protein as a viable therapeutic target, offering a potentially effective strategy for mitigating the ongoing COVID-19 pandemic (12).
The SARS-CoV-2 variations are divided into three categories: variants of interest (VOI), variants of concern (VOC), and variants under monitoring (VUM) (13). Depending on each country's circumstances, a variant's classification may change. There are five known VOCs: B.1.1.7 (United Kingdom), B.1.351 (South Africa), P.1 (Brazil), B.1.617.2 (India), and B.1.1.529 (multiple countries) (14).
This research is designed to identify SARS-CoV-2 through conventional PCR tests focused on the RdRp, E, and N genes. Additionally, the study aims to assess the genetic variations among local viral isolates compared to those worldwide.
2.1. Collection of Samples
In total, 200 nasopharyngeal samples of SARS-CoV-2 were collected randomly from September 2020 to September 2021 from the central laboratory for COVID-19 at Shahid Tahir Ali Walibag in Sulaimani, Iraq.
The samples were confirmed using variable commercial RT-qPCR kits. Subsequently, the MutaPLEX CoV-2 MUT RT-PCR kit (Immune Diagnostik, Germany, Cat. No. KG193196) was used to determine the variants.
Finally, 17 samples were randomly selected with different variants for whole RdRp, N, and E gene sequencing (sample No.1-7 Wuhan-h1, sample No. 8 and 16 Cluster 5, sample No. 9-15 Alpha (UK) B.1.1.7 and sample No. 17 Gamma P1 variant). Two samples (No. 10 and 14) were excluded because of their poor-quality Sequencing results.
2.2. RNA Extraction
According to the manufacturer's protocol, total RNA extractions were carried out using an Addprep viral nucleic acid extraction kit (AddBio Company, South Korea).
2.3. cDNA Synthesis
To prepare 50 µL of reaction mixture from each of the 17 samples, 7.5 µL from the RNA template was added to a mixture of 25 µL of master mix, 2.5 µl OligodT primers, and 2.5 µL Radom Nonamer primer and completed to 12.5 µL of Nuclease-Free H2O. The reaction mixture was then incubated under the PCR condition as follows: RNA denaturation (65°C for 5 minutes), Reverse Transcription (45°C for 50 minutes), Enzyme Deactivation (85°C for 15 minutes), and (hold at 12°C).
2.4. Conventional PCR Primers for RdRp, N and E gene
To cover the whole RdRp, N, and E gene sequence, six conserved regions were selected from the RefSeq of SARS-CoV-2 to design six sets of primers (Table 1) using Primer3 and BLAST online software. Designed Primers in silico analyzed by using NCBI Primer-BLAST to exclude miss annealing with other respiratory and coronaviruses. The primer sequences were obtained from the Reference Sequence (Wuhan Hu-1) of SARS-CoV-2 on GenBank under the accession number NC_045512.2. The lyophilized primers were then synthesized (Macrogen, South Korea).
Table 1. Primer sequences that covered the whole RdRp, N, and E gene in overlap Primer sequences
| Primers | Primer sequences (5’ to 3’) | Length |
| RdRp-1 F | TCTGTACCGTCTGCGGTATGTG | 1013bp |
| RdRp-1 R | CTTGTAGGTGGGAACACTGTAG | |
| RdRp-2 F | AACTGTTTGGATGACAGATGC | 1041 bp |
| RdRp-2 R | ACAACACGTTGTATGTTTGCG | |
| RdRp-3 F | TGATGTAGAAAACCCTCACC | 1080bp |
| RdRp-3 R | ATGACATGGTCGTAACAGCA | |
| N-1 F | ATGAGGCTGGTTCTAAATCAC | 965 bp |
| N-1 R | GCTTCTTAGAAGCCTCAGCA | |
| N-2 F | CTCATCACGTAGTCGCAACA | 955 bp |
| N-2 R | AGCTCTCCCTAGCATTGTTC | |
| E F | CGACGGTTCATCCGGAGTTG | 562bp |
| E R | CTGGCCATAACAGCCAGAGG |
In Gradient PCR, various annealing temperatures, 55°C-65°C, were employed in separate reaction wells within a thermal cycler. Simultaneously testing a temperature gradient allowed for identifying the temperature at which the specific DNA fragment amplifies efficiently. This approach serves the purpose of fine-tuning the PCR conditions specifically for the target DNA sequence of interest. Gel electrophoresis was employed to confirm the success of the PCR amplification. The temperature that yielded the most distinct and robustly amplified DNA band was determined through gel electrophoresis.
2.6. Sanger Sequencing
The PCR products, consisting of 20 μL of cDNA from the RdRp, N, and E genes, underwent sequencing analysis through the Standard-Sequencing service provided by Macrogen in South Korea.
2.7. Bioinformatics Analysis
Various tools and software were utilized in our research. Primer-BLAST facilitated primer design and specificity assessment, ApE (V3.1.3) was used for viewing and annotating DNA sequences, ClustalW (V2.0.10) for aligning multiple sequences, and Geneious Prime software (V2022.0.2) for chromatograph and text file analysis, sequence alignment, phylogenetic analysis, and generating final assemblies. The obtained sequences were submitted to GenBank for publication, securing accession numbers.
3.1. Detection of SARS-CoV-2
A total of 200 confirmed cases of COVID-19 were analyzed in this study. Among these cases, 17 samples encompassing all identified virus variants were selected for further investigation. The primer sets that were designed exhibited successful detection of the viruses in all of the tested samples, demonstrating a 100% detection rate (17/17).
3.2.Sequencing Analysis
To validate the accuracy of each sequencing result, which consisted of two reads (forward and reverse) from sample amplicons, we employed the Geneious Prime program. These final sequences were then aligned within the program using the Map to Reference alignment tool, with the reference being the SARS-CoV-2 Reference Sequence (Ref Seq) from GenBank under accession number NC_045512.2. This alignment process generated a multiple sequence alignment for the RdRp, E, and N genes.
3.3.Analysis of RdRp gene
We successfully sequenced the entire RdRp gene of SARS-CoV-2, spanning 2794 nucleotides, using three sets of primers. Among 17 tested samples, 15 exhibited optimal amplification, confirmed through agarose gel electrophoresis. Nucleotide sequence alignment of these 15 samples with the SARS-CoV-2 Reference Sequence (NC_045512.2) showed fewer mutations in Wuhan samples compared to the Alpha (B.1.1.7) and Cluster 5 (Denmark) variants. Notable mutations were found at positions 15120 C>T, 16327 A>T, and 15451 G>A in the Alpha and Cluster 5 variants, with amino acid changes observed at several positions (G59R, M196R, P223L, C643S, and C643G).
The Distances viewer revealed high similarity between some samples and the SARS-CoV-2 NC_045512.2. Three Samples (1, 6, and 7) show 100% identity with Wuhan lineage. UK variant samples (2, 3, 4, and 5) exhibited 0.01-0.03% differences from the NC_045512.2, indicating variant-specific mutations in the RdRp gene (Table 2). In summary, a heat map of differences and similarities of the SARS-CoV-2 RdRp gene by Geneious Prime software shows 100% similarity for sample NO. (1, 6, 7) and 99.9% for sample NO. (2, 3, 4, 5, 8, 11, 15, 17), 99.8% for sample NO. (9, 12, 13) and 99.7% for sample NO. (16).
Table 2. A heat map of differences and similarities of the SARS-CoV-2 RdRp gene

In the Heat Map, distances are represented by shades of gray. A lighter gray means further apart, while darker means closer together. The longest distance in the tree is white, and the shortest distance is black (may not be zero).
To describe the evolutionary relationships among the RefSeq and all 15 sample sequences, a phylogenetic tree representing a circular tree layout was built in the Geneious Prime program. The result showed the grouping of the samples based on their sequences from Contig alignment. Samples 1, 2, 3, 5, 6, and 7 were grouped as Neighbour-joining groups from the close origin as Wuhan lineage, and Samples 4 and 17 were not located in the group due to incomplete sequence assembly. Similarly, Samples 8, 9, 11, 12, 13, 15, and 16 were represented as a group obtained from Alpha lineage identified samples (Figure 1).

Figure 1. Phylogenetic analysis of RdRp gene sequences among SARS-CoV-2 by Geneious Prime software.
3.1.2. Analysis of N gene
The SARS-CoV-2 nucleocapsid gene, spanning 1259 nucleotides, was sequenced using two sets of primers, resulting in two overlapping amplicons (N1, N2). Agarose gel electrophoresis confirmed the successful amplification of each amplicon. Out of 17 mutations in the nucleocapsid phosphoprotein (N protein), nine were silent (positions: 28877-8, 28940, 28975, 29117-8, 29122, 29128, 29434, 29447, and 29518). The remaining mutations (positions: 28280-2, 28484, 28854, 28872, 28881-3, 28977, 29129, and 29131) resulted in amino acid changes, including D3L, G70S, S194L, G200D, R203K, G204R, S235F, N284K, and F285L. Wuhan samples had frequent missense mutations at positions 28884 C>T (resulting in S194L) and 28484 G>A (resulting in G70S and F285L). Alpha, Gamma, and Cluster 5 variant samples shared mutations at positions 28280-2 GAT>CTA (resulting in D3L) and 28977 C>T and 29131 T>A (resulting in S235F and F285L). Based on data analysis, some samples shared 100% similarity with the RefSeq, e.g., sample 1 (Wuhan lineage), while other samples (2-7) from the same time showed differences in sequence percentage of 0.01-0.03% from the RefSeq. UK variant samples exhibited 0.06-1.01% differences, indicating variant-specific N gene mutations (Table 3). In summary, the heatmap of differences and similarities of the SARS-CoV-2 N gene by measurement (Geneious Prime software) shows similarity of 100% for sample NO.(1),99.9% for sample NO.(4,5,7), 99.8% for sample NO.(3,6), 99.7% for sample NO.(2), 99.4% for sample NO.(8,9,15), 99.3% for sample NO.(17), 99.1% for sample NO.(11,13), 99% for sample NO.(16) and 98.9% for sample NO.(12).
Table 3. Heatmap of difference and similarities of SARS-CoV-2 N gene by measurement (Geneious Prime software)
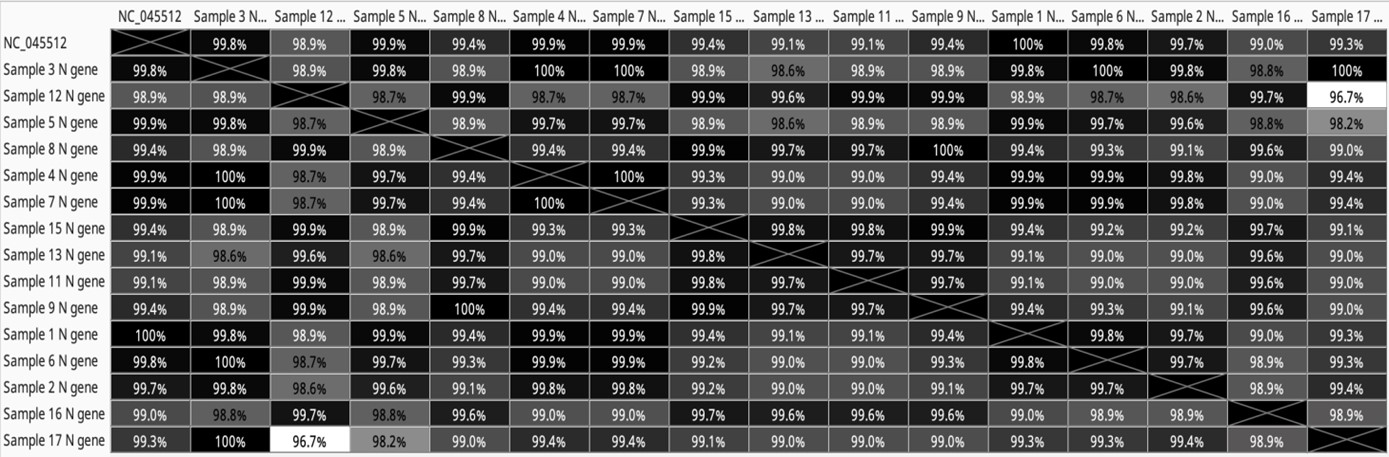
In the Heat Map, distances are represented by shades of gray. A lighter gray means further apart, while darker means closer together. The longest distance in the tree is white, and the shortest distance is black (may not be zero).
The phylogenetic result shows grouping of the samples based on their sequences from Contig alignment. Samples 1, 2, 3, 4, 5, 6, 7, and 17 were grouped as Neighbors-joining groups from close origin as Wuhan and gamma lineage. Similarly, Samples 8, 9, 11, 12, 13,15, and 16 were represented as a group obtained from Beta lineage identified samples. (Figure 2).

Figure 2. The phylogenic tree is based on the N gene whole sequences of the SARS-CoV-2 by Geneious Prime.
3.1.3 Analysis of E gene
The entire E gene sequence was obtained through cDNA preparation and Sanger sequencing, followed by Geneious Prime software analysis. Out of 15 sequences, 14 passed quality control and were assembled by mapping to the reference. Two specific samples, Sample 3 and Sample 17, exhibited mutations resulting in amino acid changes: S68F and I13L, respectively. In the analysis, most Wuhan and Alpha lineage samples shared 100% genetic characteristics with the RefSeq, except for samples 3 and 17, which showed differences of 0.04% from the RefSeq (Table 4).
Table 4. A hit map of differences and similarities of the SARS-CoV-2 E gene by measurement (Geneious Prime software)

In the Heat Map, distances are represented by shades of gray. A lighter gray means further apart, while darker means closer together. The longest distance in the tree is white, and the shortest distance is black (may not be zero).
A phylogenetic tree result showed the grouping of the samples based on their sequences from Contig alignment. All Samples were grouped as a Neighbor-joining group from the close origin of Wuhan and Alpha lineage. Similarly, except for Samples 3 and 17, they were represented as a group (Figure 3).

Figure 3. The phylogenic tree is based on the E gene whole sequences of the SARS-CoV-2 by Geneious Prime.
Upon completing the sequencing of the samples, the final results revealed that all samples exhibited the presence of the 14408C>T mutation within the RdRp coding region, the most frequently observed mutation in SARS-CoV-2. In addition to the common mutation, three lineage-specific mutations were detected in the samples. Specifically, a silent mutation occurred at position 14676C>T in samples 8, 9, 11, 12, 13, 15, and 16. Another silent mutation was identified at position 15279C>T in samples 8, 9, 11, 12, 15, and 16. Lastly, a silent mutation was observed at position 16176T>C in samples 13, 15, and 16, located in the final segment of the RdRp gene sequence.
The results showed that 9 out of the 17 mutations in N protein (position: 28877-8, 28940, 28975, 29117-8, 29122, 29128, 29434, 29447 and 29518) were silent (Table 5).
On the other hand, mutations in (position: 28280-2, 28484, 28854, 28872, 28881-3, 28977, 29129 and 29131) resulted in amino acid changes as follows (D3L, G70S, S194L, G200D, R203K, G204R, S235F, N284K and F285L) (Table 5).
In the Wuhan samples, frequent missense mutation occurred at position 28884 C>T, resulting in amino acid substitution S194L in samples No. 2, 3,4,6,7. Another missense mutation occurred in sample No.2 at position 28484 G>A and 29131 T>A, resulting in amino acid changes G70S and F285L, respectively (Table 5).
The corresponding amino acid changes at the protein level were co-occurring R203K/G204R substitutions in the N protein, detected in all alpha and cluster 5 samples (No.8-16). Another point mutation was seen at position 28280-2 GAT>CTA, substituting amino acid D3L (Table 5).
Also, two mutations were identified in Alpha, Gamma, and cluster 5 variant samples at the position (28977 C>T and 29131 T>A), resulting in the amino acid change (S235F and F285L) (Table 5). The final sequences were submitted to GenBank and published as accession numbers for whole or partial gene sequencing of RdRp 15 samples (OQ151529- OQ151543), N 15 samples (OQ253304- OQ253318) and E 14 samples (OQ253290- OQ253303).
Table 5. Analysis of SARS-CoV-2 Genome Mutations in Sulaimani, Highlighting Mutations in RdRp, N, and E Genes, Including Position, Amino Acid Substitution, Type, and Frequency per Sample.
| Mutations | Amino acid change | gene | Type of mutation | Number of isolates |
| 13975 G>C (4) | G59R | RdRp | Missense | 1 |
| 14027 T>G (3) | M196R | RdRp | Missense | 1 |
| 14408 C>T (1,2,3,4) | P323L | RdRp | Missense | 15 |
| 14676C>T (2,3) | ------- | RdRp | Silent | 7 |
| 15096 T>C (3) | ------- | RdRp | Silent | 1 |
| 15120 C>T (1) | ------- | RdRp | Silent | 1 |
| 15279 C>T (2,3) | ------- | RdRp | Silent | 6 |
| 15373 T>A (2) 15373 T>G (3) |
C643S C643G |
RdRp | Missense | 1 1 |
| 15451 G>A (1) | G671S | RdRp | Missense | 1 |
| 16092 C>T (3) | ------- | RdRp | Silent | 1 |
| 16176 T>C (2,3) | ------- | RdRp | Silent | 3 |
| 26281 A>T (4) | I13L | E | Missense | 1 |
| 26447 C>T (1) | S68F | E | Missense | 1 |
| 28280-2 GAT>CTA (2,3) | D3L | N | Point | 7 |
| 28484 G>A (1) | G70S | N | Missense | 1 |
| 28854 C>T (1,4) | S194L | N | Missense | 6 |
| 28872 G>A (3) | G200D | N | Missense | 1 |
| 28877-8 AG>TC (2) | -------- | N | Silent | 1 |
| 28881-3 GGG>AAC (2,3) | R203K G204R |
N | Point | 7 |
| 28940 C>T (2) | ------ | N | Silent | 1 |
| 28975 G>T (1) | ------ | N | Silent | 1 |
| 28977 C>T (2,3,4) | S235F | N | Missense | 8 |
| 29117-8 AC>TG (2) | ------ | N | Silent | 1 |
| 29122 A>G (3) | ------ | N | Silent | 1 |
| 29128 T>G (3) | ------ | N | Silent | 1 |
| 29129 T>G (2,3,4) | N284K | N | Missense | 4 |
| 29131T>C (1,2,3) | F285L | N | Missense | 6 |
| 29434 G>A (1) | ------- | N | Silent | 1 |
| 29447 G>T (1) | ------- | N | Silent | 1 |
| 29518 C>T (4) | ------- | N | Silent | 1 |
(1)Wuhan-hu-1, (2) Alpha B.1.1.7, (3) Cluster 5, (4) Gamma P.1
Overall, twelve mutations were recorded in eleven positions of the whole RdRp gene; six of them were silent, and another was causing a change in amino acid (Table 5).
The most prevalent mutation, 14408C>T (P323L), was identified in all 15 samples. The second most frequent mutations, 14676C>T and 15279C>T, were recorded in European variants. Notably, most RdRp gene mutations involved C>T substitutions. Phylogenetic analysis grouped Wuhan samples, except for an incomplete sequence (sample four), with European samples forming a distinct cluster.
Pachetti et al. found that the RdRp mutation, located at position 14408, present in European viral genomes starting from February 20th, 2020, is associated with a higher number of point mutations than viral genomes from Asia (16). Eskier et al., also pointed out that results indicate that the 14408C>T mutation increases the mutation rate, while the third-most common RdRp mutation, 15324C>T, has the opposite effect. It is possible that the 14408C>T mutation may have contributed to the dominance of its co-mutations in Europe and elsewhere (17). Pachetti et al., suggested that The RdRp protein structure may become rigidified as a result of the 14408C>T mutation's proline to leucine substitution (P323L), which may have an altered interaction with other replication/transcription machinery components or with other proteins (16).
The 14408C>T mutation (P323L) encodes the viral RNA-dependent RNA polymerase. This mutation was also common in KSA, as mentioned by Obeid et al. (94.9%), and was associated with severe morality (p-value < 0.0001) (18). In Italy, Boccia et al. also recorded the 14408C>T mutation (P323L) (19). Mazhari et al. showed that the P323L mutation was present in all continents North America, South America, Europe, Asia, Oceania and Africa as the mutation with the highest incidence rate (20). In the current study, the point mutation in position 14408 (C>T) was found in all samples, similar to that of Doga et al., but Maria et al. found it only in the European viral genome. On the other hand, we did not record any mutation in the position 15324C>T, as recorded by Eskier et al.
Anwar et al. studied that earlier, the most prevalent RdRp mutation in Pakistani isolates was P323L. This mutation, which is still present in the Delta variants and interacts with viral RNA in the interface domain of RdRp, stabilizes the protein. The RdRp mutation G671S found in this study has been noted as developing, present in all 18 isolates, and boosting the protein's stability (21). Kim et al. found that SARS-CoV-2 variants with RdRp mutations P323L or P323L/G671S exhibited heightened RdRp enzymatic activity at 33°C compared to 37°C, correlating with increased transmissibility in ferrets. Reverse genetics research demonstrated enhanced replication and robust transmission in ferrets for SARS-CoV-2 variants carrying RdRp mutations P323L or P323L/G671S, suggesting that these mutations contribute to increased RdRp complex stability and enzymatic activity, supporting heightened transmissibility in recent SARS-CoV-2 VOC strains (22). Both P323L and G671S were also detected in our sample No.3 as a missense mutation in position 14408C>T and 15451G>A of the RdRp gene Table (5).
Magateshvaren et al. (India) analyzed 1016 whole-genome sequences from 18 countries representing various animal hosts. Their phyloproteome study identified six major clades during the early pandemic stages, with key mutations in proteins Nsp2, Spike, RdRp, ORF3a, and N protein, including Nsp2:R218C, Nsp2:D268-(deletion), Spike:D614G, RdRp:P323L, Nsp2:A192V, ORF3a:Q57H, N protein:R203K, and N protein:G204R/L. This research emphasizes the necessity of monitoring viral evolution in non-human hosts to understand and address future pandemics (23).
Obeid et al. in KSA Studied that the 28854 C>T mutation was located in the N gene S194L. This mutation was less commonly detected (19.5%) but was still associated with higher morbidities, including death and hospitalization (18). The 28854C>T mutation resulting in the S194L amino acid substitution is identified in six samples, five of which are from Wuhan (Sample No. 2, 3, 4, 6, 7), and one is associated with the gamma variant (Sample No. 17), as indicated in Table 5.
In our study, the whole N gene of SARS-CoV-2 exhibited seventeen mutations, with nine being silent and the rest causing amino acid substitutions (Table 5). The most frequent mutation, 28977C>T (S235F), was identified in eight samples across alpha, gamma, and cluster 5 variants, previously unreported elsewhere. Two-point mutations, 28280-2GAT>CTA and 2881-3GGG>AAC, were the second most common mutations in seven samples, leading to amino acid changes (D3L, R203K, and G204R). The D3L mutation is a novel discovery, while R203K and G204R are globally recognized by the WHO as associated with increased infectivity in European variants.
Additionally, there were other missense mutations (28484G>A, 29129T>G, and 29131T>C) that caused amino acid changes at the protein level (G70S, N284K, and F285L), respectively. The first of these mutations was initially discovered in Wuhan sample No. 2, while the others were identified among the Alpha, gamma, and cluster 5 samples. Based on the information available, it is important to highlight that these three mutations have not been documented in other sources.
In Iran, evaluation of the mutations that lead to amino acid substitutions showed that SpikeD614G, RDRP-P323L, N-R203K, and N-G204R were the most frequent amino acid substitutions, as mentioned by Yavarian et al. (24). Boccia et al, from Italy also record 28881-3 GGG>AAC mutations (R203K and G204R) (19). Tsuchiya et al. from Japan observed 395 amino acid changes in the N protein, where R203K and G204R were the most widespread mutations. In B.1.1, which was the most frequently detected lineage during the latter period of the first wave, four amino acid substitutions were commonly identified: S: D614G, N: R203K, N: G204R, and RdRp: P323L (25). Sia et al. observed that the two N protein mutations R203K and G204R, which have been found in the alpha variation B.1.1.7, gamma variant P.1, lambda variant C.37, and omicron variant BA.1/B.1.1.529, would be harmful (26).
Our study detected a mutation in position 28881-3GGG>AAC in European samples. Five alpha variants were sampled: 9,11,12,13,15, and two clusters were 5 samples: No. 8 and 16. This point mutation causes amino acid substitution (R203K and G204R). Azad Gajendra revealed that Arg-92 is mutated to Serine, which might affect N protein and RNA interactions (27). All SARS-CoV-2 variants of interest or concern defined by the WHO contain at least one mutation with >50% penetrance within seven amino acids (N: 199–205) in the nucleocapsid (N) protein, which is required for replication and RNA binding, packaging, stabilization and release (28).
Syed et al. tested 15 N mutations, including two combinations corresponding to the Alpha and Gamma variants since they both contain the co-occurring R203K/G204R mutations. The Alpha and Gamma variant N improved luciferase expression in receiver cells by 7.5- and 4.2-fold, respectively, relative to the ancestral Wuhan-Hu-1 N-protein. In addition, four single amino acid changes improved luciferase expression: P199L, S202R, R203K, and R203M (29). Wu et al. found that the R203K/G204R mutations are adaptive and linked to the high-transmissibility SARS-CoV-2 lineage B.1.1.7. These mutations confer a replication advantage, potentially related to enhanced ribonucleocapsid assembly. The 203K/204R virus demonstrates increased infectivity in human lung cells and hamsters. The study observes a positive correlation between COVID-19 severity and 203K/204R frequency, suggesting their contribution to heightened transmission and virulence in select SARS-CoV-2 variants. Additionally, mutations in the nucleocapsid protein, alongside those in the spike protein, play a crucial role in viral spreading during the pandemic (30).
Reports on thorough mutational analysis from the GISAID database show that 2% of strains are E mutants. Higher amino acid changes in the SARS-Cov-2 E protein's C-terminal domain, including S55F, V62F, and R69I, could, however, affect how the protein binds to tight junction proteins, which could impact pathogenesis (31).
In the current study, mutations in two samples (17, 3) cause amino acid changes in I13L and S68F. Our recent analysis shows that the mutation at position 26281 A>T in Wuhan sample 3 leads to an amino acid substitution (I13L). This finding has not been reported in any other sources.
Rizwan et al. identified significant associations between amino acid substitutions in the E gene (T9I, F4F, F4L, P71L, V49L, L65L, P71S, L73L, S68F, and R69I) and disease severity, with S68F linked explicitly to the severity of disease in the gamma variant (sample No. 17) (32). Cao et al. demonstrated that the E protein plays a crucial role in virus packaging and reproduction, and its deletion weakens or abolishes virulence (33). A recurring mutation at position 26340G>U in the SARS-CoV-2 E gene is associated with E gene failure in the cobas system by Roche (6). Abavisani et al. found that the E protein's most frequent mutation is T9I, varying regionally, with T9I prevalent in Europe, Oceania, North America, and South America. At the same time, P71L is most common in Africa and Asia. V62F ranks among the top 10 mutations in Asia, Europe, and North America but is less prevalent in Africa, Oceania, and South America (34).
Nakhaie et al. from Kerman, Iran, detailed that SARS-CoV-2 variant classification entails the taxonomy of different strains or variants of the virus based on specific genetic mutations or alterations within its genome. These variants undergo identification through genomic sequencing and analytical procedures, facilitating an enhanced understanding of the dissemination patterns, evolutionary trajectories, and potential impacts of distinct viral lineages (35).
Using conventional PCR techniques, our study successfully designed six primer sets tailored for precise amplification of SARS-CoV-2 genes, with a specific focus on RdRp, N, and E. Utilizing sequencing analysis, we observed a gradual increase in mutation rates over time. The most frequently identified mutation was 28977C>T (resulting in the S235F amino acid substitution), found in eight samples associated with the alpha, gamma, and cluster 5 variants. Notably, this mutation has not been reported in any other documented sources to date.
Additionally, we identified a novel point mutation, 28280-2GAT>CTA, resulting in the amino acid change D3L. Three mutations (28484G>A, 29129T>G, and 29131T>C) leading to amino acid substitutions (G70S, N284K, and F285L) were also discovered, with no prior reports in the existing literature. Furthermore, the mutation at position 26281 A>T in the Wuhan variant, resulting in an amino acid substitution (I13L), is documented for the first time and has not been found in any other sources, based on our current study.
Understanding mutations in the RdRp, N, and E genes of SARS-CoV-2 is imperative for gaining insights into the virus's biology diagnostic liability, for example, selecting the E gene, formulating effective therapeutic strategies, and proactively addressing potential challenges posed by viral evolution.
We want to thank the Kurdistan Regional Government, the Ministry of Higher Education and Scientific Research, and the Sulaimani University presidency.
Ethics approval
The Ethics Committee of the Department of Biology, College of Science, University of Sulaimai, Iraq, approved the research on 19 September 2023 (protocol code UoS-Sci-Bio 0010).
Conflicts of Interest
The authors declared no conflict of interest.
Conceptualization, S.A; methodology, S.A. and D.A; software, Z.H.; validation, S.A., D.A. and Z.H.; formal analysis, D.A. and Z.H; investigation, D.A.; resources, D.A and Z.H.; data curation, S.A.; writing—original draft preparation, D.A.; writing—review and editing, S.A and Z.H.; visualization, D.A.; supervision, S.A. All authors have read and agreed to the published version of the manuscript.
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Received: 2023/08/18 | Accepted: 2023/11/20 | ePublished: 2023/11/29
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
Copyright Policy
Iranian Journal of Medical Microbiology by Farname is licensed under CC BY-NC 4.0![]()
![]()
![]()
