




![]()
![]()
![]()
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
URL: http://ijmm.ir/article-1-1390-en.html



2- Razi Herbal Medicines Research Center, Lorestan University of Medical Sciences, Khorramabad, Iran
3- Department of Obstetrics and Gynecology, Lorestan University of Medical Sciences, Khorramabad, Iran
4- Department of Medical Biotechnology, Faculty of Medicine, Lorestan University of Medical Sciences, Khorramabad, Iran , pejman7genetian@gmail.com
Coronaviridae is a family of viruses consist of seven members that have a single-stranded RNA with positive-sense, enveloped, and possess genome size of 17 to 91 kb (1). Four viruses from this family cause the common cold, and two of them lead to the deadly respiratory disease of severe acute respiratory syndrome (SARS) and middle east respiratory syndrome (MERS). The newly discovered seventh member of this family causes COVID-19 disease (2).
Based on genotyping and serology, coronaviruses are divided into alpha, beta, gamma and delta genus that alpha and beta genus caused health problems in human (3). In late 2019, a new beta coronavirus was isolated from people exposed to a seafood market in Wuhan, China, and named SARS-CoV-2 (4). Symptoms such as fever or chills, cough, shortness of breath or difficulty breathing, fatigue, muscle or body aches, headache, the new loss of taste or smell, sore throat, congestion or runny nose, nausea or vomiting, digestive problems, diarrhea, kidney failure among the main symptoms of COVID-19 (5-8).
SARS-CoV-2 spread globally, and the World Health Organization (WHO) and the Public Health Emergency of International Concern (PHEIC) declared a pandemic (9). After pandemic waves from Wuhan strain, a rapidly spreading variant emerged in the UK. This variant is derived from the SARS-CoV-2 20B/GR clade (lineage B.1.1.7). It contains multiple mutations, including a combination of the N501Y and the 69–70del in Spike gene. These mutations caused increased transmissibility of the virus up to 71% over and above the previous circulating strains (10). Although a number of vaccines have been introduced to the community, it remains necessary to design a vaccine using conserve viral antigens, which cover current circulating variants.
SARS-CoV-2 possesses 14 open reading frames in its genome, including four structural proteins: the Spike protein (S), the Envelope protein (E), the membrane protein (M), and the Nucleocapsid protein (N) (11, 12). Among these structural proteins, Spike proteins that reside on the virus surface are typically selected as antigens to produce antibodies from B-cells to neutralize the virus. Spike protein contains the receptor-binding domain (RBD) responsible for binding the ACE2 (angiotensin-converting enzyme 2) and entry into the cell. Therefore, the conserved sequence of S protein is a major target antigen for vaccine development (13-17).
Nucleocapsid protein (N) plays an important role in the viral cell cycle and contributes to forming helical ribonucleoproteins during the RNA genome packaging, regulate viral RNA synthesis during replication, transcription, and modulating metabolism in infected individuals. On the other hand, N gene is more conserved and stable. It is proved that high-level IgG against this protein secreted in sera of SARS patients, and the N protein is a representative antigen for the T-cell response in a vaccine setting (18, 19). The immune system, by its nature, can make its own adjustments to recognize vaccines and pathogens. Low immunogenicity is one of the drawbacks of vaccines. One approach to overcome this problem is to use the proper adjuvant (20).
Today, SARS-COV-2 mutations are one of the challenges in human populations immunization, so designing a vaccine that can target the conserved sequence of the virus is a new approach in this study. In the current study, with the aim of in silico immunoinformatic, a novel and common epitope of S and N proteins from COVID-19 significant variants predicted for designing multi-epitope vaccine against this global health problem.
2.1. Retrieval of Protein Sequences and Alignment
The FASTA format of surface glycoprotein (RefSeq: YP_009724390.1) and Nucleocapsid phosphoprotein (RefSeq: YP_009724397.2) of SARS-CoV-2 Wuhan strain were obtained from the National Center of Biotechnology Information NCBI at: https://www.ncbi.nlm.nih.gov/protein/?term=SARS-CoV-2 (21).
Multiple sequence alignment was performed through CLUSTALW at: https://www.genome.jp/tools-bin/clustalw
2.2. B-cell epitope (linear) prediction
The Immune Epitope Database (IEDB) server at: http://tools.iedb.org/bcell/ includes data about immune epitopes for immune response system purposes. Bepipred linear epitope prediction server at http://www.cbs.dtu.dk/services/BepiPred/ was applied for predicting linear B cell epitopes based on target antigens-specific sequence features using amino acid and HMM scales (22, 23).
2.3. Prediction of HLA Class I and II Epitopes
RANKEPEP server at: http://imed.med.ucm.es/Tools/rankpep.html using Position Specific Scoring Matrices (PSSMs), Class I and Class II MHC molecules predicts the sequence of target antigens (24).
2.4. Allergenicity, Toxicity and Antigenicity Prediction of the Selected Epitopes
The AllerTOP server at: https://www.ddg-pharmfac.net/AllerTOP/ is a bioinformatics tool for predicting allergens. This server is the first server alignment-free that can predict whether or not target antigens are allergenic (25).
Server ToxinPred at: http://crdd.osdd.net/raghava/toxinpred/ is one of the best bioinformatics tools for predicting the toxicity of target antigens. The server for the detection from toxic epitopes of machine-learning technique support vector machine (SVM) is used (26).
Server VaxiJen at:
http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html is an in silico tool for predicting the antigenicity of target peptides. This server is the first server alignment-independent that can predict whether or not target peptides are protective antigens (27).
2.5. Generation of the 3D structures of the selected epitopes
The PEP-FOLD 3 server at: https://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD3/ is based on the structural description of peptides. In a short time, this server returns useful data in five top models.
2.6. Molecular docking and refinement of the selected epitopes
PatchDock web servers at: https://bioinfo3d.cs.tau.ac.il/PatchDock/php.php are applied for molecular docking. This server is applied carry out structure prediction of protein–small and protein–protein molecule collections (28). Investigation of molecular docking of peptides using this server against alleles HLA DRB1*04-01 (PDB ID: 5JLZ) and HLA-A*11-01 allele (PDB ID: 5WJL) was done. PatchDock server algorithm is based on shape complementarity principles. This server uses the root mean square deviation (RMSD) score for candidate solutions. Top score solutions are considered the best solutions by the PatchDock server. In order to check the refinement and re-scoring of the docking results were carried out, we used FireDock server at: http://bioinfo3d.cs.tau.ac.il/FireDock/php.php
The server generates global energy for the best solutions and produces them based on global energy, and the lowest global energy is always considered the best docking score (29).
2.7. Construction of final vaccine construct
The potential epitopes of the two target antigens were selected and used in the final construction structure. After selecting the final epitopes, it is time to merge the epitopes into each other through the appropriate linkers GSGSGS, EAAAK and AAYKK. In this study, Maltese-bound protein (MBP) was used as adjuvants for the designed vaccine. This sequence is recognized by Toll Like Receptors (especially TLR2 and TLR4) and stimulates cellular and humoral immunity (30).
2.8. Evaluation of Antigenicity and allergenicity of recombinant vaccine
The antigenicity of the recombinant construction was specified using the ANTIGENpro server at: http://scratch.proteomics.ics.uci.edu/.
The allergenicity of the recombinant construction was determined using AllergenFP v1.0 server at: http://ddg-pharmfac.net/AllergenFP/.
2.9. Analysis of physicochemical properties of recombinant vaccines
In current study, ProtParam server at: https://web.expasy.org/protparam/ was used for calculating physicochemical properties of the novel vaccine, including molecular weight, theoretical pI (isoelectric point, EI (extinction coefficient), R and +R (total number of positive and negative residues), instability index II, GRAVY (grand average hydropathy) and AI (aliphatic index) (31).
2.10. Analysis of the secondary and tertiary structure of the recombinant vaccine
To predict the secondary structure, the GOR4 server at:
https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_gor4.html was used. This server predicts the data theory of the second structure of the novel vaccine.
The three-dimensional structure of recombinant construct was predicted by Phyre2 web server at: http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index. Phyre2 server provides a useful tool to analyze and predict protein structure and function. This server is one of the best protein online prediction servers and works based on the hidden Markov model (HMM) (32). YASARA software was used for predicted models’ visualization.
2.11. Refinement and Validation of the Third Structure of the Recombinant Vaccine
In order to check the refinement of modeled 3D structure, we used GalaxyRefine at: http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE. In the refined model, two servers, including RAMPAGE and ProsA, were used for validation.
RAMPAGE server at: http://mordred.bioc.cam.ac.uk/~rapper/rampage.php calculate phi–psi torsion angles for each amino acid in the vaccine construct (33).
ProSA-web server at: https://prosa.services.came.sbg.ac.at/prosa.php applied for structure validation and calculated an overall quality score (34).
2.12. Molecular Docking Study
Docking experiments were performed by the Z-Dock server (http://zdock.umassmed.edu/). This server carries out the prediction of interactive protein-protein docking (35). The Cluspro server (https://cluspro.bu.edu/login.php) was used to evaluate the interaction between the recombinant vaccine and TLR4 / MD-2. On this server, vaccine protein was selected as ligand, and TLR4 / MD2 was introduced as a receptor for molecular docking (36).
2.14. Molecular Dynamics Simulation
We used the iMODS server at: http://imods.chaconlab.org/ to check the stability of the protein complex. This server is a useful tool for molecular dynamics study. This server was used to explain the motion of a collective protein in internal coordinates through normal mode analysis (NMA). The server estimated the direction and extent of the immanent motions of the complex in terms of deformability, eigenvalues, B-factors, and covariance (37).
2.14. Codon Adaptation and in Silico Cloning
SMS server at: https://www.bioinformatics.org/sms/rev_comp.html was used for reverse translation of the designed construct to the nucleotide sequence. In the next step, the Java Codon Adaptation tool (http://www.jcat.de/) was used for codon optimization to clone designed vaccine in Escherichia coli K12 strain expression host. Restriction sites BamHI and HindIII were introduced at the N and C-terminal sites of the final construct for cloning in the pET 26+ expression vector.
All steps taken to design a recombinant vaccine are shown in Figure 1.
3.1. B-cell Binding Epitopes
Surface glycoprotein and nucleocapsid phosphoprotein sequences were used for B-cell binding epitopes prediction by Immune Epitope Database (IEDB) server (Table 1).
Table 1. B-cell peptide candidates
| ANTIGENS | POS | SEQUENCE | SCORE |
| Spike protein | 251 | PGDSSSG | 1.484 |
| 807 | PDPSKPS | 1.413 | |
| 252 | GDSSSGW | 1.404 | |
| 476 | GSTPCNG | 1.397 | |
| Nucleocapsid protein |
2 | SDNGPQN | 1.439 |
| 18 | GGPSDST | 1.417 | |
| 75 | NTNSSPD | 1.417 | |
| 196 | NSTPGSS | 1.413 |
3.2. T-cell Binding Epitopes
Surface glycoprotein and nucleocapsid phosphoprotein sequences were used for MHC I and MHC II binding epitopes prediction by the RANKEPEP server (Tables 2, 3).
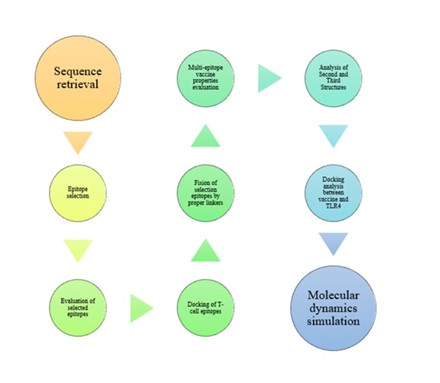
Figure 1. Schematic procedure chart for designing the recombinant vaccine. The procedure used for designing the recombinant vaccine has briefly been shown.
Tables 2. MHC-I peptide candidates
| ANTIGENS | POS | SEQUENCE | SCORE |
| Spike protein | 154 | ESEFRVYSS | 19.406 |
| 196 | NIDGYFKIY | 16.09 | |
| 661 | ECDIPIGAG | 13.93 | |
| 576 | VRDPQTLEI | 13.088 | |
| 1180 | QKEIDRLNE | 12.734 | |
| Nucleocapsid protein |
295 | GTDYKHWPQ | 15.609 |
| 103 | DLSPRWYFY | 13.047 | |
| 397 | AADLDDFSK | 11.648 | |
| 272 | QAFGRRGPE | 7.955 |
Table 3. MHC-II peptide candidates
| ANTIGENS | POS | SEQUENCE | SCORE |
| Spike protein | 144 | YYHKNNKSW | 19.71 |
| 199 | GYFKIYSKH | 17.658 | |
| 1208 | QYIKWPWYI | 17.142 | |
| 832 | GFIKQYGDC | 16.276 | |
| 169 | EYVSQPFLM | 15.561 | |
| 955 | NAQALNTLV | 15.311 | |
| 896 | FAMQMAYRF | 13.314 | |
| 365 | YSVLYNSAS | 12.812 | |
| Nucleocapsid protein |
86 | YYRRATRRI | 27.321 |
| 85 | GYYRRATRR | 20.179 | |
| 385 | RQKKQQTVT | 12.064 | |
| 387 | KKQQTVTLL | 11.317 | |
| 237 | KGQQQQGQT | 10.056 |
3.3. Allergenicity, Toxicity and Antigenicity determination
T-cell and B-cell epitopes that were selected by RANKEPEP and IEDB servers have been evaluated for allergenicity, toxicity, and antigenicity by mentioned servers in the material and method section (Tables 4). In the next step, ESEFRVYSS, VRDPQTLEI, DLSPRWYFY and QAFGRRGPPE epitopes were selected from MHC Class I, and EYVSQPFLM, FAMQMAYRF and RQKKQQTVT epitopes were selected from MHC Class II and PGDSSSG, GDSSSGW and GSTPCNG epitopes were selected from B-cell.
Table 4. Allergenicity, antigenicity, and toxicity analysis of the selected T-cell and B-cell epitopes of Spike protein and Nucleocapsid protein.
| ANTIGENS | EPITOPES | ANTIGENCITY | ALLERGENCITY | TOXICITY |
|---|---|---|---|---|
| Spike protein | PGDSSSG | ANTIGEN | NON-ALLERGEN | NON-TOXIN |
| PDPSKPS | NON-ANTIGEN | NON-ALLERGEN | NON-TOXIN | |
| GDSSSGW | ANTIGEN | NON-ALLERGEN | NON-TOXIN | |
| GSTPCNG | ANTIGEN | NON-ALLERGEN | NON-TOXIN | |
| ESEFRVYSS | ANTIGEN | NON-ALLERGEN | NON-TOXIN | |
| NIDGYFKIY | NON-ANTIGEN | ALLERGEN | NON-TOXIN | |
| ECDIPIGAG | ANTIGEN | ALLERGEN | NON-TOXIN | |
| VRDPQTLEI | ANTIGEN | NON-ALLERGEN | NON-TOXIN | |
| QKEIDRLNE | NON-ANTIGEN | NON-ALLERGEN | NON-TOXIN | |
| YYHKNNKSW | NON-ANTIGEN | NON-ALLERGEN | NON-TOXIN | |
| GYFKIYSKH | NON-ANTIGEN | ALLERGEN | NON-TOXIN | |
| QYIKWPWYI | ANTIGEN | ALLERGEN | NON-TOXIN | |
| GFIKQYGDC | NON-ANTIGEN | ALLERGEN | NON-TOXIN | |
| EYVSQPFLM | ANTIGEN | NON-ALLERGEN | NON-TOXIN | |
| NAQALNTLV | ANTIGEN | ALLERGEN | NON-TOXIN | |
| FAMQMAYRF | ANTIGEN | NON-ALLERGEN | NON-TOXIN | |
| YSVLYNSAS | NON-ANTIGEN | ALLERGEN | NON-TOXIN | |
| Nucleocapsid protein |
SDNGPQN | ANTIGEN | NON-ALLERGEN | NON-TOXIN |
| GGPSDST | ANTIGEN | NON-ALLERGEN | NON-TOXIN | |
| NTNSSPD | ANTIGEN | NON-ALLERGEN | NON-TOXIN | |
| NSTPGSS | ANTIGEN | NON-ALLERGEN | NON-TOXIN | |
| GTDYKHWPQ | NON-ANTIGEN | ALLERGEN | NON-TOXIN | |
| DLSPRWYFY | ANTIGEN | NON-ALLERGEN | NON-TOXIN | |
| AADLDDFSK | NON-ANTIGEN | NON-ALLERGEN | NON-TOXIN | |
| QAFGRRGPE | ANTIGEN | NON-ALLERGEN | NON-TOXIN | |
| YYRRATRRI | NON-ANTIGEN | NON-ALLERGEN | NON-TOXIN | |
| GYYRRATRR | NON-ANTIGEN | NON-ALLERGEN | NON-TOXIN | |
| RQKKQQTVT | ANTIGEN | NON-ALLERGEN | NON-TOXIN | |
| KKQQTVTLL | NON-ANTIGEN | NON-ALLERGEN | NON-TOXIN | |
| KGQQQQGQT | ANTIGEN | ALLERGEN | NON-TOXIN |
3.4. Generation of Three-dimensional Structures of Epitopes and Molecular Docking
In this study, molecular docking was used to determine whether the final epitopes were able to bind to MHC class I and class II. For docking study, selected MHC class 1 and 2 epitopes were introduced to the server as ligand, and the HLA-A*11-01 allele (PDB ID: 5WJL) and HLA DRB1*04-01 (PDB ID: 5JLZ) were presented as the receptor. Amongst MHC-I and MHC-II epitopes from surface glycoprotein, ESEFRVYSS and EYVSQPFLM demonstrate the outcome with the lowest and best global energy score. Amongst MHC-I and MHC-II epitopes of nucleocapsid phosphoprotein, DLSPRWYFY and RQKKQQTVT demonstrate the best outcome with the lowest and highest global energy score (Table 5).
Table 5. Results of molecular docking for T-cell epitope against two HLA types
| Name of the protein | Epitope | MHC allele | Global energy | Hydrogen bond energy |
| Spike protein | ESEFRVYSS | HLA-A*11-01 | -29.75 | -2.56 |
| VRDPQTLEI | HLA-A*11-01 | -22.86 | -2.18 | |
| EYVSQPFLM | HLA DRB1*04-01 | -18.27 | -4.95 | |
| FAMQMAYRF | HLA DRB1*04-01 | -7.96 | -2.12 | |
| Nucleocapsid protein |
DLSPRWYFY | HLA-A*11-01 | -48.03 | -2.83 |
| QAFGRRGPE | HLA-A*11-01 | -9.09 | -1.31 | |
| RQKKQQTVT | HLA DRB1*04-01 | -16.99 | -1.31 |
3.5. Epitope Selection and Construction of the Multi-epitope Peptide Vaccine
Based on high-ranking T-cell and B-cell epitopes, fourteen epitopes from two antigens were selected as the final regions. The final epitopes of each antigen were fused together by GSGSGS and AAYKK linkers. Maltose / maltodextrin-binding protein was also added as an adjuvant to the N-terminal of the novel vaccine with Linker EAAAK. The designed novel vaccine construct consisted of 590 amino acid residues, as illustrated in Figure 2.

Figure 2. a) Graphical illustration of the multi-epitope vaccine construct. b) The sequence comprises 590 amino acid sequences. The vaccine contains an MBP adjuvant at the N-terminal of vaccine construct
3.6. Evaluation of Antigenicity and Allergenicity of Recombinant Vaccine Construct
The results of the AllergenFP 1.0 server showed that the designed structure was not an allergen. The possibility of the designed recombinant construct antigenicity was predicted 0.909 by the ANTIGENpro server, which means that our novel vaccine can stimulate impressive immune system responses.
3.7. Analysis of Physicochemical Properties of Recombinant Vaccines Construct
The physicochemical parameters of the recombinant construct include GRAVY, theoretical pI, half-life, instability index, aliphatic index, amino acid composition, and molecular weight are shown in Table 6.
Table 6. Prediction results of recombinant vaccine physicochemical parameters
| Physicochemical properties | Result |
| GRAVY | -0.453 |
| theoretical pI | 5.50 |
| half-life | The estimated half-life is: 30 hours (mammalian reticulocytes, in vitro). >20 hours (yeast, in vivo). >10 hours (Escherichia coli, in vivo). |
| instability index | 26.44 |
| aliphatic index | 63.27 |
| Total number of negatively charged residues | 64 |
| Total number of positively charged residues | 58 |
| molecular weight | 61854.64 |
3.8. Secondary and Tertiary Structure Analysis
GOR IV server predictions show that our recombinant vaccine was composed of 33.90% alpha helix (H), 15.08% extended strand, and 51.02% random coil (C) secondary structural elements. Also, the three-dimensional model of our novel vaccine is produced by the phyre2 server and is shown in Figure 3. The Phyre2 web server showed three main domains in the third structure of the designed vaccine. This server indicated that 369 residues (63% of vaccine sequence) had been modeled with 100.0% confidence by the single highest scoring template.

Figure 3. The predicted 3D structures of the recombinant vaccine generated by the Pyre2 server. 3D structure of protein vaccine structures generated by YASARA software.
3.9. Tertiary Structure Refinement and Validation
The selected model was refined using GalaxyRefine. This server introduced five refined three-dimensional models. The first model with the highest score was selected and displayed in Figure 4.

Figure 4. Final vaccine construct after refinement via GalaxyRefine server. The server genertaed five refined three-dimensional models. The first model with the highest score was selected and visulized by YASARA software.
The refined novel vaccine structures were validated with the aid of the ProSA-web and RAMPAGE servers. The ramachandran plot server showed that the recombinant vaccine had 99.2% number of residues in the favour region and 0.8% number of residues in the allowed region (Figure 5a). ProSA-web servers were used for the evaluation of potential and quality errors in the 3D crude model. This server is utilized for the prediction of Z-score prediction, which is found as −11.91 (Figure 5b).
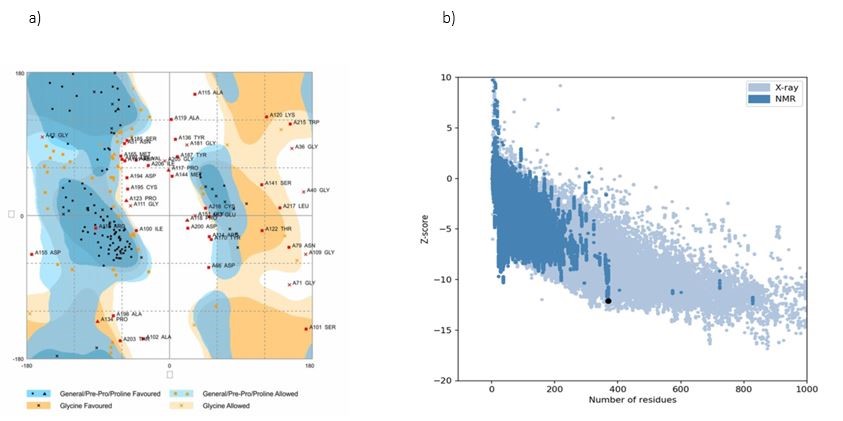
Figure 5. a) Ramachandran plot generated by RAMPAGE server. Ramachandran diagram showed that the recombinant vaccine had 99.2% number of residues in the favored region and 0.8% number of residues in the allowed region. b) ProSA-web server was used for the evaluation of potential and quality errors in 3D crude model. The Z-score of the constructed model calculated −11.91, which is in the range of native protein conformation scores.
3.10. Molecular Docking of Subunit Vaccine with TLR-4
A novel multi epitopes vaccine was docked with TLR4 using the Z-DOCK server. In general, ten complexes have been developed, and the most appropriate set of TLR vaccines has been selected based on the correct combination and binding (Figure 6a). The Cluspro server was also used to evaluate the interaction between the recombinant vaccine and TLR4 / MD-2. The top 10 models were selected based on the biophysical characteristics of the receptor and ligand, which in terms of the docking pattern between the receptor and the ligand, the first introduced model possess the lowest weight score (-915.9 kcal / mol) (Figure 6b).

Figure 6. Docking studies of designed multi-epitope vaccine and modeled TLR4. a) Docked complex of TLR4 and multi-epitope vaccine generated by Z-DOCK server. b) Cluspro server used to generate binding sites of multi-epitope vaccine with TLR4.
3.11. Molecular Dynamic Simulation
Natural state analysis (NMA), large-scale flexibility, and stabilization of the recombinant vaccine were investigated by the iMOD server. This server is relevant to the internal coordinates of the docked complex. Figure 7a shows the probable deformity of the vaccine-TLR-4 related to the individual distortion of any residue shown by the chain hinges (green). Figure 7b shows the B-factor values equivalent to the RMS inferred via NMA. Figure 7c shows the eigenvalue for the complex, equal to 1.922. Figure 7d demonstrated the colored bars of the cumulative (green) and individual (red) variances that are inversely related to the eigenvalue. Figure 7e shows the coupling between the different interactions of the residues by the covariance matrix, i.e., uncorrelated to white, red-correlated, and anti-correlation with blue. Figure 7f shows the elastic network model that distinguishes pairs of atoms associated with springs.

Figure 7. Schematic displaying the findings of MD simulation study of TLR-4 and recombinant vaccine docked complex. Here, (a) eigenvalues, (b) B-factor, (c) deformability, (d) variances, (e) covariance and (f) elastic network analysis.
3.12. Codon Optimization and in silico Cloning
The SMS server conducted back translation of the vaccine construct. JCAT server was used to assess the key properties of the vaccine DNA sequence, including the GC content and Codon Adaptation Index (CAI). The CAI value for the optimized nucleotide sequence was 1, and the GC content of the optimized sequence was calculated as 50.11% in E. coli. These outcomes suggest that the optimized vaccine DNA sequence will have a maximum expression yield in E. coli.
The number of people who get infected by COVID–19 diseases has increased day by day, as well as the increase in mortality that has caused international concern (38). Therefore, vaccination is an effective way to prevent COVID–19 diseases (39).
In this research, we determined the epitopes of potentially immune cells of two SARS-CoV-2 antigens and designed a multi-epitopes vaccine against current variants COVID-19. The SARS-CoV-2 antigenic proteins were evaluated by using in-silico tools, and then the antigenic parts of the Nucleocapsid and Spike proteins were selected for use in the novel vaccine. Immunoinformatic approaches are novel strategies for identifying, designing, and manufacturing specific antigen epitopes for use in a variety of purposes such as; vaccines, immunotherapy, etc. (40).
The novel multi-epitope vaccines are designed to stimulate the immunity of specific pathogens by selectively stimulating specific B-cell and T-cell antigens (41). In comparison with viruses that posse’s DNA genetic material, viruses that contain RNA genetic material such as SARS-CoV-2 have a greater tendency to mutation. These mutations in this virus occur because of the lack of proofreading activity of polymerases, and this phenomenon causes resistance to drugs and escapes from immune surveillance (42-44). These mutations led to emerging of new variants, of which the UK B1.1.7 strain is one. This strain possesses transmissibility of up to 71% over, increased clinical severity of illness, and vaccine escape capability (10).
Spike protein has two important regions with conserved sequences, include the receptor-binding domain (RBD) and N-terminal domain (NTD). These two regions are characterized by surface exposure and high antigenicity, and these two regions are outside of the area prone to mutations (45). In contrast, the Nucleocapsid protein is more conserved and stable, with 90% amino acid homology and fewer mutations over time. Nucleocapsid proteins of many coronaviruses are expressed abundantly during infection and are highly immunogenic (46). It was reported that high levels of IgG antibodies against N protein were secreted in the sera of SARS patients (47). In SARS vaccine design, N protein was used for T-cell proliferation, response, and cytotoxic activity (48, 49).
The COVID–19 multi-epitope vaccine design was performed by in silico tools in several steps, includes immunoinformatic analysis, selection of conserved epitopes and multi-epitopes vaccine construction, immunoinformatic evaluation, analysis of secondary and third structures and study of the interaction between the novel vaccine and ligand (TLR-4). An effective vaccine must have both T-cell epitope and B-cell epitope to stimulate cellular and humoral immunity against pathogens (41). Appropriate servers were used to predict epitopes of T-cells and B cells. There are two main types of cells that have functions in the immune system, including T lymphocytes and B lymphocytes (50). The epitopes of T-cell (MHC-I, MHC-II) and B-cell were identified by the servers of RANKEPEP and IEDB, respectively. High-scoring and potential epitopes (T-cell and B-cell epitopes) with non-toxicity, non-allergenicity, and antigenicity were selected for recombinant vaccine construction.
Then, the PEP-FOLD server generates three-dimensional structures of MHC class-I epitopes and MHC class-II epitopes to study molecular docking. All selected T-cell epitopes have acceptable molecular docking scores, so it can be concluded that all selected epitopes have the ability to bind to their respective targets and stimulate an immune response in a robust manner.
After studying the docking of T-cell epitopes, the design of the recombinant construction was performed. An adjuvant is an essential component of the novel human vaccines (51). These compounds are used to increase the immunogenicity of vaccines, including DNA vaccines and multi-epitope vaccines (52). Therefore, combining adjuvants with protein epitopes can effectively stimulate specific antigen-specific immune responses (53). MBP, a protein with a molecular weight of 42 kDa, is responsible for binding and transporting maltose from the periplasmic space of bacteria of the Gram-negative model. MBP has been reported to act as an adjunct to specific immunity through specific receptors. It has also been reported that this protein increases lymphocytes (54-56). In this recombinant vaccine construct, MBP was used as an adjuvant. On the other hand, MBP also can facilitate the expression and purification process of recombinant vaccine in E. coli, by one-step purification using affinity chromatography (57).
The linker is an important component in fusion epitopes to the design of a recombinant construction. Based on the structure of the linkers, the linkers are divided into 3 types, which include: flexible linkers, rigid linkers, and cleavable linkers. Hare, GSGSGS flexible linker and AAYKK cleavable linker were used for fusion between the epitopes, and the EAAAK rigid linker was used for fusion between the adjuvant and the epitope (58).
After vaccine construct design, the allergenicity, toxicity, antigenicity, and physiochemical analysis revealed that the designed vaccine was stable and possessed proper immunogenicity. Moreover, molecular docking and molecular dynamics simulations indicate that the designed vaccine has stable interaction with TLR4 ligand.
In the end, codon adaptation and in silico cloning experiments were performed to ensure that vaccine efficiently express in the target host, E. coli strain K12. (59, 60).
COVID-19 pandemic is the deadliest outbreak in recent decades. Prevention of the newly emerging novel coronavirus infection and mutated form of that is a very challenging problem for global health. In silico tools can help us to conquer this problem by saving time and cost. In this study, immunoinformatic and reverse vaccinology help us design a potential multi-epitope vaccine against this new virus. Various in silico and computational studies of the proposed vaccine constructs indicate that this multi-epitope vaccine might confer proper immunogenic response. With complementary experimental and clinical studies, the current study should be helpful to researchers to develop a potential vaccine against SARS-CoV-2 virus variants.
This research was funded by the Deputy of Research and Technology, Lorestan University of Medical Sciences, Khorramabad, Iran (grant NO. 1397-1-99-1345). The authors appreciate this financial support.
Conflicts of Interest
All authors declared that there is no conflict of interest.
Funding
This research was financially supported by Lorestan University of Medical Sciences, Khorramabad, Iran. Hereby the authors appreciate all the people who helped in this research.
Ethics approval
The present study was approved by The Ethics Committee of Lorestan University of Medical Sciences (IR.LUMS.REC.1398.293). This article does not contain any studies with human participants or animals performed by any of the authors.
Received: 2021/07/11 | Accepted: 2021/09/1 | ePublished: 2021/09/28
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
Copyright Policy
Iranian Journal of Medical Microbiology by Farname is licensed under CC BY-NC 4.0![]()
![]()
![]()
