




![]()
![]()
![]()
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
URL: http://ijmm.ir/article-1-1384-en.html



2- Molecular and Medicine Research Center, Arak University of Medical Sciences, Arak, Iran
3- Department of Medical Biotechnology, Arak University of Medical Sciences, Arak, Iran
4- Infectious Diseases Research Center (IDRC), Arak University of Medical Sciences, Arak, Iran
5- Molecular and Medicine Research Center, Arak University of Medical Sciences, Arak, Iran ,
Sexually transmitted infections (STI) can be caused by pathogens that have been acquired through sexual activity (1). STIs are the common cause of illness worldwide, and they represent a significant economic, therapeutic and social problem (2, 3). The number of patients with STIs is higher than the one reported in the statistics because of the asymptomatic infection and the absence of partners testing. Several bacteria can lead to these infections. Chlamydia trachomatis and Mycoplasma genitalium are two common causes of STIs. STIs can cause cervicitis, pelvic inflammatory disease, endometritis, salpingitis, spontaneous abor-tion, and prematurity/low birth weight (4, 5). From 1996, about 20% of the patients suffered from Chlamydia infections annually, and in the United States, every year, up to 4 million people develop Chlamydia infections (6).
Mycoplasma genitalium is one of the critical species of Mycoplasma, which is isolated from the reproductive system. This bacterium has no peptidoglycan (PG) wall, has a minimal genome, and is a non-gonococcal -urethritis agent, pelvic inflammatory disease, and spontaneous abortion (7, 8). Most infections in 70-80% of cases are indistinguishable because they are often asymptomatic and become chronic if not treated. Therefore, an efficient diagnostic method is needed due to the rapid outbreak of infection and the increase in STI-related illnesses and their treatment costs (9).
There are many laboratory methods to detect C. trachomatis and M. genitalium, ranging from cell culture to amplification tests. In addition, in the case of antibiotic treatment, the patient has false-negative results. On the other hand, it will take about 72 hours and has low analytical specificity and sensitivity (10, 11).
Serologic tests are based on antigen identification, direct fluorescent antibody (DFA), and enzyme-linked immunosorbent assay (ELISA), but also have low sensitivity than molecular method (70-85%) (11).
Recently it has been proven that molecular meth-ods, in particular, polymerase chain reaction (PCR) and real-time PCR, are helpful for identifying pathogens (13-15). In addition, compared to PCR, real-time PCR has lower environmental pollution, higher sensitivity and more accurate results (2, 12, 13).
Although PCR methods have efficiency, rapid results, and higher sensitivity and specificity than other standard microbiological methods, they are not cost-effective (18, 19). In developing countries, rising prices for commercial kits hinder the widespread use of these tests. The use of in-house appliances and the multiplex format has overcome the price problem, made this test feasible, and can be used as an applied test (14). So this study aimed to develop a TaqMan Duplex real-time qualitative PCR method for rapid detection, with high sensitivity and specificity of both bacterial C .trachomatis and M. genitalium.
Samples
We used samples of DNA bank ofmolecular and medical research center; (Arak, Iran) for molecular detection. Samples were 110 suspected cases of sexually transmitted diseases from women with high-risk behaviors with STIs from the Center for promotion and women's health, Arak city, during 5 months in 2016. Samples were taken from the endocervical and vaginal swabs transmitted through the transfer medium phosphate buffer saline (PBS) to the laboratory and analyzed by real-time PCR molecular technique for the presence of C. trachomatis and M. genitalium bacteria.
Primers and Probe Designing
For two bacterial agents of STI, the particular genomic sequences from the NCBI GenBank database were subjected to multiple alignments to identify conserved sequences to design probe and primer.
Due to the lack of access to the standard strains of these bacteria and the need for positive control in the reaction, as well as the design of the primer and probe, all sequences related to the conserved region of the ompA gene (GeneBank ID: 884473) in C. trachomatis and the MgPa gene (GeneBank: GU2261-99.1) in the M. genitalium were extracted from the NCBI website and compared with each other. Alignm-ent was done with ClustalW by Mega4.0so-ftware (Center for Evolutionary Functional Genomics. Tempes, AZ). Taqman probes and primers were designed with Taxa specific/cross-species. The align-ment of primers and probes was done using Mega4.0, and they were designed using AlleleIDso-ftware (Premier BiosoftInternational, Palo Alto, CA,-USA).The fluorophore of the end of the ompA probe of the FAM molecule and the MgPa fluorophore probe of the Cy5 molecule are considered. The end quenchers of the two probes were the BBQ (Black Hole Quencher) molecule. The sequences of the primers and the TaqMan probes (Table 1) were ordered to Copenhagen Company (Denmark).
Furthermore, according to the designed primers and probes were selected the most conserved area from each gene and ordered the cloning of the pBSK (+) plasmid including the synthesized sequence of OmpA of C. trachomatis and MgPa of M. genitalium from Gene Synthesis Services (Biomatik CO. Canada) as a positive control in real-time PCR. Negative control also was water instead of DNA as a template. In addition, the primer was designed to amplify a 501 bp region of the human beta-2 microglobulin (B2M) gene (ID: 567)as an internal control (IC).
Real-time PCR assays
Genomic DNA was extracted from endocervical and vaginal samples using a DNA mini kit (QIAamp, Qiagen, Hiden, Germany). DNA was eluted in 50 µL of elution buffer. The real-time PCR was performed in a light cycler 96 thermocycler (Roche Diagnostics-,Germany).Duplex amplification was performed in 20µL volume including 5 µL of extracted 30ng DNA,10µ of 2X mastermix (YTA, Iran),0.3 µL probe 20pmolOmpA and 0.3 µL probe 20pmol MgPa, 0.6 µL sense and anti-senseprimers10pmolof OmpA and 0.8 µL sense and anti-senseprimers of 10pmol MgPa. The amplification profile contained a single cycle of polymerase activation at 95°C for 10 min, followed by 40 cycles of denaturation at 95 for 10s, annealing at 58°C for 10 s, extension at 72°C for 15 s.
Table 1. The designed primers for the Duplex Real-Time PCR assay
| ID | Sequence 5/-3/ | Oligonucleotide size | Ampliconsize (bp) |
|---|---|---|---|
| C.trachomatis | |||
| Sense primer | 5-TGAGTTCTGCTTCCTCCTTGC-3` | 21 | 135 |
| Anti sense primer | 5`-TGATAGCGTCACACCAAGTGG-3 | 21 | |
| Anti sense probe | 5`-TCTCCRCCRAAACCTTCCCAYAGA-3` | 24 | |
| M.genitalium | |||
| Sense primer | 5-GATTGATCACCATCCYAATGAAG-3` | 23 | 145 |
| Anti sense primer | 5`-ATATAGTAAAGTAGCAGCCACTGG-3` | 24 | |
| Sense probe | 5`-TGGTATGGTCCACTTCGCCACTGT-3` | 24 | |
| B2M | |||
| Sense primer | 5`- TCCATCCGACATTGAAGTTG-3` | 20 | 501 |
| Anti sense primer | 5`- GAAGCCAGTAGGTAAGAAGTG-3` | 21 | |
At first, the reactions were performed in singleplex mode and optimized the determination of annealing temperature and Mgcl2 concentration. Then the duplex reactions were optimized with primers and probe mixtures and finally by adding specific internal control primers. Components and items during PCR reactions were optimized, including concentration and ratio of two pairs of primers, annealing tem-perature, time of annealing and extension, the concentration of positive control genes, and the number of cycles. The sensitivity and diagnostic specificity of the tests were calculated basis of Medical statistical calculators, specificity= true negative /(true negative + false positive) and sensitivity= true positive /(true positive + false negative).
In addition, for all samples, real-time PCR was performed using the Chlamydia Trachomatis/Ureap-lasma/M. genitalium Real-TMcommercial kit (Sacace, Italy). In brief, 5μL of PCR-mix-1, 2.5μL of PCR-mix-2, and 0.25μLTaqF Polymerase enzyme were added to the microtube per sample. Finally, 0.1 μL of the above mixtures were added to tubes of Light Cycler 96, and 1 μL of extracted DNA of samples was added to each tube. The temperature profile of the kit was followed: The enzyme activation at 95°C for 10 min, followed by 5 cycles of denaturation at 95 for 10s, annealing at 60°C for 20 s, extension at 72°C for 15 s and finally 20 cycles of denaturation at 95 for 5s, annealing at 60°C for 30 s, extension at 72°C for 15 s.
Duplex Real-time PCR Reaction
The product length of amplified fragments for OmpA, MgPam, and B2M were 135 bp, 145bp, and 501 bp, respectively. The PCR reactions were optimi-zed, performed in single and duplex format, and ana-lyzed on a 1% agarose gel electrophoresis (Figure 1).
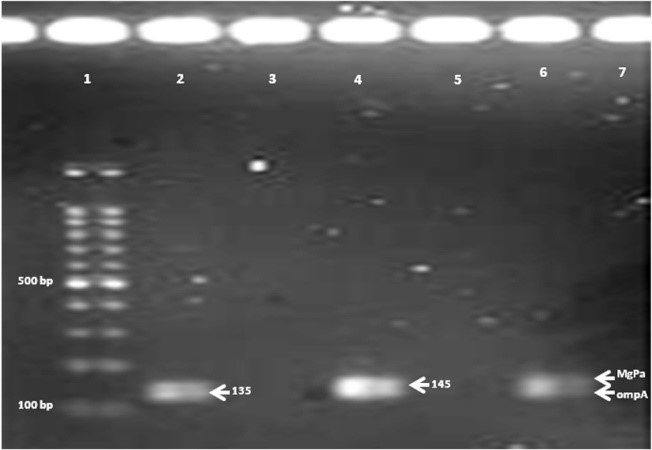
Figure 1. Agarose gel of amplicons of ompA C. trachomatis {135bp} and MgPa M. genitalium {145 bp} in single {lans 2 and 4} and duplex PCR {lane 6}. Lanes 3, 5 and 7 are negative controls.
Two primers related to each gene were mixed individually and duplexed and then used at different temperatures in the PCR reaction to investigate the best annealing temperature. Electrophoresis results showed the best temperature was 58 degrees centigrade with the highest color intensity.
The BLAST result of the designed primers showed that the primers of each bacterium identified specifically only the desired bacterium. Therefore, in silico analysis, the primers were specific for C. trachomatis and M. genitalium and IC.
To ensure that primers do not react with other bacteria that may cause the STIs and for determination of specificity of the test, the PCR test with bacterial DNA strains purchased from the Center for the Collection of Fungi and Microbes, results showed was done without any reaction with bacteria of the list as mentioned above (Table 2).
Table 2. The list of negative control bacteria
| IROST* ID | Bacteria | Row |
| PTCC 1621 | Serratia marcescens |
|
| PTCC 1768 | Streptococcus agalactiae |
|
| PTCC 1683 | Streptococcus mutans |
|
| PTCC 1600 | Citrobacter freundiii |
|
| PTCC 1776 | Proteus mirabilis |
|
| PTCC 1221 | Enterobacteraerogenes |
|
| ا PTCC 1402 | Klebsiellaoxytoca |
|
| PTCC 1440 | Staphylococcus saprophyticus |
|
| ATCC 25923 | Staphylococcusaureus |
|
| PTCC 1435 | Staphylococcus epidermidis |
|
| PTCC 1240 | Streptococcus pneumoniae |
|
| PTCC 1760 | Neisseriameningitidis |
|
| PTCC 1766 | Haemophilus influenzae |
|
* Iranian Research Organization for Science and Technology
Fluorescence signals due to C. trachomatis proliferation with fluorescence probes labeled with FAM dyes and M. genitalium proliferation labeled with Cy5C probes in the LightCycler®96 were shown in Figure 2.
To optimize the concentration and ratio of primers, different amounts of primers with intervals0.1 (concentration from0.1 to0.8 μM) were evaluated several times until the concentration was at the lowest Cq and the lowest primer dimers creation. The experiment was performed as a duplex reaction for each sample. Results were analyzed in red and blue channels. The presence of B2M as IC indicated the accuracy of all previous steps, including DNA extraction, etc. Also, internal control primers at low concentrations and different sizes of PCR products led to amplification of the internal control not interfering with main products.
Analytical Specificity
The analytical specificity of the method was determined evaluating the cross-reactivity of primers, assessing homologies to all sequences deposited in the NCBI databank and using the nucleotide BLAST algorithm. Also, we evaluated the specificity panel by some potentially cross-reactive pathogens. The results revealed no amplification from genomes from these pathogens was detected.
Diagnostic Evaluation
The experiments showed that according to the developed duplex assay of a total of 110 endocervical samples collected from the women promotion and the health center, 9 samples had positive results. Thus, 8 samples (7.2%) were positive for C. trachomatis and 1 sample (0.9%) for M. genitalium. All 110 samples were re-evaluated by Chlamydia trachomatis / Ureaplasma / M. genitalium Real-TM commercial kit (Table 3). The evaluation was performed with positive control and negative control by the kit mentioned earlier.
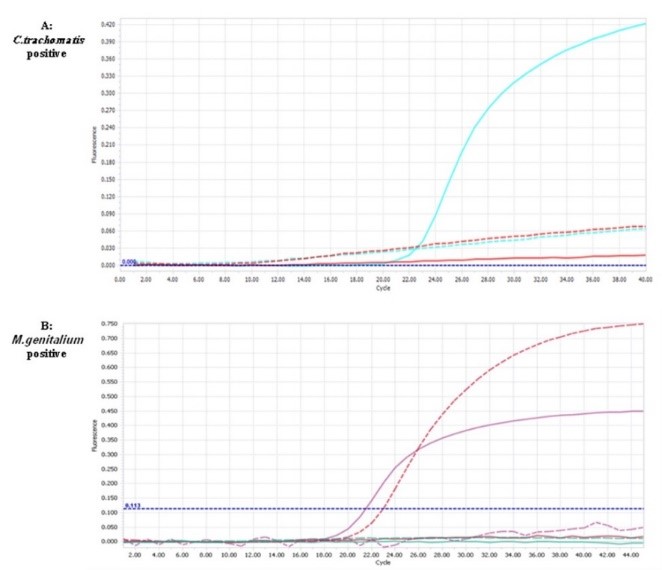
Figure 2. The amplification curves of the positive sample of control C. trachomatis in FAM channel, cq 23.2 (A) and M. genitaliumin channel Cy5C, cq 21.5 (B). {Cq: quantitation cycle}
Table 3. Comparison of results obtained with the duplex real-time PCR and C. trachomatis /Ureaplasma/ M. genitalium Real-TM
| Chlamydia trachomatis/Urea plasma/M.genitaliumReal-TM | ||||
| Total | - | + | Duplex Real-Time PCR | |
| 9 | 3 | 6 | + | |
| 97 | 94 | 3 | - | |
| 106 | 97 | 9 | Total | |
In the used commercial kit study, 5 of the 8 positive samples of C. trachomatis were positive and in addition to 1 positive M. genitalium, 3 other samples also showed positive results. However, using the kit method, 3 samples of C. trachomatis were not positive compared to the designed duplex method, and 3 more samples of M. genitalium had positive results. The sensitivity and specificity of in-house protocol for C. trachomatis were 100% and 97%, respectively, specificity and sensitivity of in-house protocol for M. genitalium were 100% and 57%, respectively, and finally, the diagnostic sensitivity of the duplex assay was 75%, and the diagnostic specificity of the duplex assay was 97.22%.
STIs are one of the critical causes of the disease worldwide, and their quick diagnosis is essential. Although other detection methods such as culture and serology are available to detect STIs, it seems molecular methods are more sensitive and specific (2, 15). Real-time PCR is the most sensitive method derived from PCR and the most potent quantification method in research and diagnostic laboratories. In addition, it was performed faster than PCR and without electrophoresis, and the contamination rate was lower. There are some methods of the real-time PCR assay for simultaneous detection of infections that causes sexually transmitted diseases. The most important disadvantages of commercial kitsin developing countriesare their high cost, mainly when performed on a single-plex (23-27). In the present study, a duplex Real-time PCR was performed to detect bacterial pathogenic and sexually transmitted diseases in vaginal endocervical samples. In our method, all two PCR reactions are carried out simultaneously in a tube, reducing the cost of materials and the reaction components of the Real-Time PCR.
Smith and his colleagues evaluated a multiplex PCR assay to detect STI agents (C. trachomatis and M.genitalium). The sensitivity and specificity of this diagnostic assay were 97% and 85%. Compared to the present study, duplex Real-time PCR is more sensitive and corresponds to the given research (28).
The designed primers of the present research were taxa specific for conserved regions of the genome and capable of identifying all particular strains of these bacteria that were reported sequences, up to the date of designing. Also, none of the primer pairs were cross-reactive, which was confirmed by the analytical specificity of the duplex assay. In optimizing multiplex PCR methods, designing specific primers for infectious agents is essential. Thus, primers should have several general characteristics, including the lack of cross-reactivity with other agents, the proximity of all temperature melting of primers, and production of different sizes so that these products can be easily distinguished from each other on the gel. In addition, we used the Real-TM commercial kit as a molecular reference method to evaluate the diagnostic accuracy of the duplex Real-time PCR method (Table 3).
The main problem of in-house methods is that they mainly have no validation process for the clinic. Therefore, this research aimed to design and use a multiplex PCR method that is capable was sensitive and fast to detecting C. trachomatis bacteria and M. genitalium in the endocervical specimen. In the present study, primer sets are designed taxa-specific and therefore considered all reported sequences, up to the date of designing, from target genes of any bacteria.
A study by Wei et al.in China in 2012 performed multiplex real-time PCR for simultaneous identification of C. trachomatis and Ureaplasma parvum with 63 cervical specimens collected from patients with non-gonococcal urethritis. They studied 64 samples; 8 were Chlamydia positive, 20 were Ureaplasma positive, and 11 were positive for both organisms (29). Their method is consistent with our study.
Using the developed method, 9 samples had positive results, which were confirmed in at least 6 cases using a valid commercial kit. Using the designed method, 8 samples of C. trachomatis and 1 sample of M. genitalium were positive. Compared with the kit method, 1 sample of Chlamydia and 3 samples of Mycoplasma were positive, which is consistent with the results of many previous studies and shows the importance of using molecular methods to diagnose STIs. On the other hand, the comparison of the results obtained from the designed methods and the commercial kit showed that the sensitivity and specificity of the duplex test are 75% and 97%, respectively. However, when using clinical specimens, the test's sensitivity was reduced to 75% due to the inability of the designed method to detect 3 specimens with infectious agents (M.genitalium) who had previously been commercially positive by commercial real-time PCR.
Our designed method was not able to detect 3 samples of M. genitalium positive and had 3 contradictory samples in the case of C. trachomatis, as two leading theories are hypothesized. Including these 3 positive samples of Chlamydia are probably considered false-positive results, and the diagnostic feature of the designed method is reduced to 70%. In addition, the commercial kit mastermix is standard and high quality; due to the cost-effectiveness of our developed in-house approach, the used mastermix had a lower grade, which could be a reason for the reduced sensitivity of the developed test. Also, due to the completion of the student project and the prevalence of COVID-19, it is impossible to collect more samples for this paper.
The detection of STIs using conventional microbiological methods is complicated and time-consuming (30). Although the sensitivity of the duplex method was lower than the commercial kit, by changing the color of the fluorescence of the CY5 probe, the determined sensitivity is also well-responding to the diagnostic requirement of endocervical samples, which are suspected of STI.
Also, the designed method has advantages over conventional PCR. For instance, the number of pollution samples was significantly reduced due to their closure lid in the microtube, and a large number of samples were analyzed in a short time. Another advantage of the designed method is that it is entirely duplex, and all two PCR reactions are performed simultaneously in a tube. This feature reduces material consumption and real-time PCR reaction components. We propose to provide more validation methods for the production kits in future projects. Regarding kit production, the sensitivity and specificity of this test should be increased by using better quality reaction mixtures and utilizing control materials. The biggest challenge in the present study was the lack of sufficient financial resources, which led to the use of materials and reaction mixtures with lower quality, as well as a lower number of clinical samples.
The analysis of the results showed that the specificity and sensitivity of the kits were higher. Nevertheless, the advantages of designing in-house protocols for commercial kits are their low cost. The developed duplex RT method in this study can simultaneously diagnose C. trachomatis and M.genita-lium. In addition, a comparison of this method with a valid commercial way showed that this method could replace commercial methods for detecting common causes of bacterial STI.
This study was supported by an MSc grant to the first author from Arak University of Medical Sciences (Ethics committee ID: 93-174-12). We are thankful to the office of research and technology of Arak University of Medical Sciences.
Conflicts of Interest
The authors declare that they have no conflict of interest.
STI: Sexually transmitted infections
C. trachomatis: Chlamydia trachomatis
M. genitalium: Mycoplasma genitalium
PG: peptidoglycan
NAAT: nucleic acid amplification tests
DFA: direct fluorescent antibody
ELISA: enzyme-linked immunosorbent assay
PCR: Polymerase Chain Reaction
B2M: beta-2 microglobulin.
Received: 2021/07/7 | Accepted: 2021/10/9 | ePublished: 2022/01/20
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
Copyright Policy
Iranian Journal of Medical Microbiology by Farname is licensed under CC BY-NC 4.0![]()
![]()
![]()
