




![]()
![]()
![]()
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
URL: http://ijmm.ir/article-1-1596-en.html


2- Department of Medical Biotechnology, School of Medicine, Lorestan University of Medical Sciences, Khorramabad, Iran
3- Department of Medical Biotechnology, School of Medicine, Lorestan University of Medical Sciences, Khorramabad, Iran , pejman7genetian@gmail.com
Leishmaniasis is one of the most important infectious diseases, and zoonosis caused by Leishmania parasites and is the most important tropical disease after malaria (1, 2). Leishmaniasis is one of the primary transferable global health issues in many tropical and subtropical countries and it is endemic in 102 countries (3-5). According to the World Health Organization (WHO) report, more than 350 million people are at risk of leishmaniasis infection. Annually 200,000-400,000 people have visceral leishmaniasis (VL) and 700,000-1200,000 people have cutaneous leishmaniasis (CL). The disease has now spread to some non-endemic regions. The disease burden (daily) is reported to be 3.3 million people; clinical manifestations are cutaneous (CL), cutaneous mucosal (MCL), and visceral (VL) (6-10).
Cutaneous leishmaniasis (CL) is the most common form of leishmaniasis, transmitted to humans by biting a female sandfly. One of the causes of this disease is the intracellular parasite Leishmania major (11, 12). Cutaneous leishmaniasis causes ugly ulcers that remain in place for a long time. It also leaves scars after healing and, in terms of beauty and psychological effects on the patient, causes major problems (13, 14).
The only treatments available for cutaneous leishmaniasis are pentavalent antimonials, amphotericin B, and pentamidine. However, drug-resistant and drug toxicity are two serious concerns, which inevitably limit the chemotherapeutic options. Hence, preventive strategies such as vaccines may seem more useful in combating cutaneous leishmaniasis (15). Unfortunately, despite all the efforts made by applying different vaccination strategies, no vaccine is available to humans available against leishmaniasis (16-18).
Scientists have studied the production of various vaccines, such as a killed parasite, subunit, and DNA (19). A small number of vaccines available to combat Leishmania parasites have entered the clinical phase, so many studies are needed to find a way to treat leishmaniasis (13).
The most important factor in designing a strong vaccine for CL, is identifying the host immunity against L. major (20). During CL infection, the leishmaniasis parasite enters the macrophage of an infected person, activating the Th1 and Th2 responses. Th1 responses produce interferon-gamma and IL-2. Th2 responses produce antibodies and cytokines IL-10 and IL-4 (21).
One of the significant factors in immunization is that there are epitopes in vaccination that the immune system recognizes. The epitope is the fragment of the antigen which is detected by the immune system. Therefore, in the structure of novel vaccines (Multiepitope peptide vaccines), the identification of immunogenic epitopes could be very practical (22, 23).
Bioinformatics tools have boosted the recognition of the epitopes (24). Bioinformatics is modern science that uses computers, computer software, and databases to answer biological problems, especially in the cellular and molecular fields. Bioinformatics is an online tool based on various databases and algorithms, which is created and applied to predict protein structures, cellular and molecular properties, epitopes, and more (25, 26).
Identification of effective antigens and appropriate adjuvants are able to increase the potency and immunogenicity of novel vaccines (27, 28).
In this study, LACK, CPB, and KMP-11 proteins, which are candidate proteins for the CL vaccine, were selected for the study.
Kinetoplastid membrane protein 11 (KMP11) is a complex protein highly associated with Leishmania promastigotes lipophosphoglycan. Lipophos-phoglycan is strongly antigenic to human T cells. KMP11 is a protein specific to Kinetoplastida, capable of building innate and adaptive immunity against Leishmania. Among the different Leishmania molecules known as possible vaccine antigens, KMP-11 has attracted much attention due to its high human T cell antigen (29-31).
Another antigen is LACK (Leishmania homologue of receptors for Activated C Kinase), which plays a prominent role in the immunopathogenesis of Leishmania major by generating a Th2 response. T cells produce IL-4 against LACK antigen, resulting in resistance to leishmaniasis infection. The use of this protein as a DNA vaccine in mice was immunogenic and protective (32).
Another selective antigen is a cysteine protease involved in developing parasitic diseases and survival, host cell infection, and escaping from the host immune system; therefore, it has been suggested as a target therapy molecule. Cysteine proteases are considered pathogens of Leishmania parasites. Protein derived from cysteine protease B is involved in modulating immune system activities such as inducing IL-4 production, inhibiting the production of IL-12 by macrophages, and analyzing MHC-II molecules (33, 34).
Multiepitope peptide vaccines (Novel vaccines) designed by bioinformatics tools not only cope with a variety of pathogens effectively but also reduce the negative effects of irrelevant immune sequences (35). Despite all the benefits of using a multiepitope peptide vaccine, the prime issue with these vaccines is a low immune promotion (36). To solve the problem, adjuvants are recommended (37).
Toll-like receptors (TLRs) are protected receptors that are expressed in immune and non-immune cells and are known as a class of pattern recognition receptors (PRRs); those specific molecules recognize pathogens as agonists. Two TLR4 agonists, RpfE and RpfB, have been used as adjuvants to enhance vaccine safety. Various studies have used them as effective adjuvants in vaccines (38, 39).
Novel vaccines (Multiepitope peptide vaccines) include protected B and T cell epitopes, which can be a useful approach for the advancement of vaccines for infectious diseases. Studies have been performed to design multiepitope peptide vaccines for disease agents that have shown their ability to provide immunity against these agents (40-42).
We attempted to identify and anticipate the best and most successful functional B and T cell epitopes for immunization in this work using bioinformatics technologies. The resuscitation-promoting factor RpfE and RpfB of Mycobacterium TB are two TLR4 agonists that were used as adjuvants in this study. In addition, we focused on producing a recombinant multiepitope vaccine that contained effective antigenic epitopes.
The data illustrated the structure of the recombinant multiepitope vaccine was not allergic and could evoke humoral and cellular immune responses. The vaccine structure based on the bioinformatics system, which is reported here, provides significant immunogenic potential that may be further evaluated in the next phase of in vivo and in vitro study.
2.1. Protein Sequence Retrieval
Protein sequences of candidate antigens related to LACK (Accession no. AAB88300.1), CPB (Accession no. AUL80104.1), and KMP-11 (Accession no. AAR84616.1) were attained from the National Centre for Biotechnology Information (https://www.ncbi.nlm.nih.gov/) (43).
Mycobacterium TB RpfE and RpfB were obtained from UniProt at www.uniprot.org and used as TLR4 agonists. RpfE (Entry: 053177) and RpfB (Entry: 053177) (Entry: P9WG29, G5 domain).
2.2. Evaluation of Antigen-determining Characteristics
ProtParam server was used for physicochemical characterization of 3 selected antigens like molecular weight, amino acid composition, half-life, aliphatic index, theoretical pI, GRAVY, and so on (https://web.expasy.org/protparam/) (44).
Furthermore, the antigenicity of every protein was shown using the ANTIGENpro server. This server (http://scratch.proteomics.ics.uci.edu/) is a pathogen -independent predictor with an accuracy of 82 percent. (45).
The SOPMA (Self-Optimized Prediction Method with Alignment) secondary structure analysis tool was used to predict four states: helix, beta-sheet, coil, and turn (https://npsa-prabi.ibcp.fr/NPSA/npsa_sopma.html) (46).
2.3. Anticipation of MHC-II Epitopes
MHC-II binding epitopes were selected through RANKPEP server. Human MHC-II alleles, which are HLA-DRB 1101, HLA-DRB 0401, and HLA-DRB 1 101 were selected to predict the MHC-II binding epitope.
RANKPEP server predicts MHCII epitopes based on position-specific scoring matrices (PSSMs). The binding threshold for the highest epitope scores is 4-6% MHC-II (http://imed.med.ucm.es/Tools/rankpep.html) (47).
2.4. Prediction of MHC-I Epitopes
The MHC-I epitope analysis was assessed using the IEDB server. HLA-A * 01: 01 and HLA-B * 07: 02 were among the alleles used to predict MHC-I binding. Artificial Neural Networks (ANN), Stabilized Matrix Approaches (SMM), and Score Matrices derived from Combinatorial Peptide Libraries may all be employed as estimation methods. The consensus technique (https://www.iedb.org/) is employed, a mix of multiple approaches like ANN, SMM, and other combine the algorithms (48, 49).
2.5. Prediction of B-Cell Epitopes
The BCpred server was applied for linear anticipation of B cell epitopes. The BCpred server uses a Subsequence String Kernel (SSK) and Support Vector Machine (SVM) with 74.54% and 75% accuracy and specificity, respectively (https://webs.iiitd.edu.in/raghava/bcepred/) (50).
2.6. Prediction of CTL Epitopes
The CTLPred service was used to anticipate cytotoxic T lymphocyte (CTL) epitopes. This server employs a direct approach incorporating quantitative matrix (QM) and machine learning approaches. In addition, the server employs a mix of methodologies and consensus estimation. The consensus method technique was used to anticipate the default cut-off score (0.00) (http://www.webs.iiitd.edu.in/raghava/ctlpred/index.html). (51).
2.7. Anticipation of Interferon-Gamma Induction Epitopes
Interferon gamma-induced epitopes of MHC-II binding epitopes are predicted to design a useful multiepitope recombinant vaccine. The IFNepitope server uses a database made of IFN-gamma-induced and non-induced MHCII. IFNepitope server also uses various methods, including machine-learning, motif-based search, and a hybrid approach. The maximum accuracy attained from the hybrid model is 81.39% (https://webs.iiitd.edu.in/raghava/ifnepitope/index.php) (52).
2.8. Design of a Recombinant Vaccine
To create a recombinant multiepitope vaccine, the results obtained from the predicted epitopes were compared, and the epitopes with the highest and common scores were selected. Selected epitopes can evoke different sorts of immune responses. Selected epitopes were combined with adjuvants by a suitable linker.
2.9. Evaluation of the Characteristics of the Recombinant Multi-epitope Vaccine
The antigenic and antigenic responses of the recombinant vaccine construct were assessed using the Vaxijen v2.0 server. Vaxijen v2.0 server predicts antigenicity antigenic response based on target organism antigens such as bacterial, viral, and tumoral antigens.
This server employs a cutting-edge, alignment-independent method for predicting antigenic reaction probability. Its target organism accuracy ranges from 70% to 85% (http://www.ddgpharmfac.net/vaxijen/VaxiJen/VaxiJen.html) (53).
The AllerTop v.2.0 server was used to estimate the allergenicity of the recombinant vaccination. With an accuracy of 85.3 percent, this site predicts allergies and non-allergies using the K-Nearest Neighbors (KNN) algorithm
(http://www.ddg-pharmfac.net/AllerTOP) (54).
The physicochemical characteristics of the recombinant multiepitope vaccination were assessed using a ProtParam server. In vitro and in vivo half-life, theoretical pI, molecular weight (MW), amino acid composition, instability index, in vitro and in vivo half-life, aliphatic index, and Grand average of hydropathicity index (GRAVY), and molecular weight (MW) were among the physicochemical parameters calculated (https://web.expasy.org/protparam/) (44).
2.10. Secondary Structure Evaluation
The server GOR IV, secondary structure (alpha-helix, beta-sheet, turn, or random coil) was analyzed (https://npsaprabi.ibcp.fr/cgi-bin/secpred_gor4.pl) (53).
2.11. Homogeneity Modeling
The third structure of the recombinant multiepitope vaccine was predicted by utilizing the I-TASSER server. The predicted three-dimensional models are known as confidence scores, known as C scores (higher values indicate higher confidence) (http://zhanglab.ccmb.med.umich.edu/I-TASSER/ download/) (54).
2.12. Refinement of 3D Model Structure
GalaxyRefine performed the refining process of the 3D model. According to C score, the best 3D model from I-TASSER has been processed in GalaxyRefine server. GalaxyRefine is likely to improve the early models (http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE) (55).
2.13. Validation of the Refined Third Structure
The 3D manufacture of the ultimate vaccine has been verified using ProSAweb, Verify3D, Rampage, and ERRAT servers.
Using RAMPAGE's phi-psi torsion angles, RAMPAGE divides the protein into three groups: preferred outlier areas, permitted regions, and outlier regions. (http://mordred.bioc.cam.ac.uk/~rapper/rampage.php) (56).
ProSAweb calculates the total quality score for a particular input structure. The server simply needs Cα atoms to evaluate low-resolution structures. Highlighting errors in experimental and theoretical models of protein structures is a great issue in structural biology.
ProSA-web is a web-based interface to the ProSA software, which is often used to validate protein structures. For a given input structure, ProSA produces an overall quality score. If this rating falls outside of a typical range for natural proteins, the design is likely to have mistaken. (https://prosa.services.came.sbg.ac.at/prosa.php) (57).
Non-bonded atomic interactions allow the ERRAT server to discriminate between proper and erroneous protein structure regions.
(http://servi ces.mbi.ucla.edu) (58).
A three-dimensional protein model was tested for compliance with the vaccine's sequence using Verify3D to validate its three-dimensional structure. (http://services.mbi.ucla.edu/) (59).
2.14. In silico Cloning
The Sequence Manipulation Suite server (https://www.bioinformatics.org/sms/) was used to conduct reverse translation and codon optimization of vaccine protein sequences.
Another method used to investigate sequence properties was the GenScript Rare Codon Analysis Tool, which can be found at https://www.genscript.com/tools/rare-codon-analysis (https://www.genscript.com/tools/rare-codon-analysis) (CFD).
Eventually, restriction sites HindIII and BamHI were introduced to the N and C terminal sequences of the ultimate vaccine's DNA to clone it into the E. coli vector pET-14b.
3.1. Retrieve and Collect Sequences
In the present study, LACK, CPB, and KMP-11 proteins were selected as antigenic determinants. Sequences for three antigenic peptides were retrieved in the FASTA format at the NCBI site. The amino acid sequences for RpfE and RpfB were obtained in FASTA format from the UniProt dataset. RpfB is the most important part of linker to TLR4 (60).
3.2. Analysis of Antigen-determining Structures
The physicochemical properties, secondary structure analysis, and antigenicity prediction of all three proteins were determined and summarized in Table 1.
Table 1. Analysis of selective antigens
| LACK | KMP-11 | CPB | Features evaluation of antigen determinants |
| 312 | 92 | 349 | Number of amino acids |
| 34.42 KD | 11.23 KD | 37.95 KD | Molecular weight |
| 6.05 | 5.96 | 7.09 | Theoretical pI |
| 36 | 20 | 31 | Total number of negatively charged residues (Asp + Glu) |
| 30 | 17 | 31 | Total number of positively charged residues (Arg + Lys) |
| >10 hours | >10 hours | >10 hours | Estimated half-life (Escherichia coli, invivo) |
| 31.38 | 50.02 | 35.65 | Instability index |
| 80.61 | 33.04 | 77.48 | Aliphatic index |
| -0.275 | -1.447 | -0.095 | Grand average of hydropathicity (GRAVY) |
| 13 is 4.17% | 77 is 83.70% | 117 is 33.52% | Alpha helix |
| 136 is 43.59% | 0 is 0.00% | 63 is 18.05% | Extended strand |
| 47 is 15.06% | 2 is 2.17% | 19 is 5.44% | Beta turn |
| 116 is 37.18% | 13 is 14.13% | 150 is 42.98% | Random coil |
| 0.751214 | 0.456471 | 0.775114 | Predicted Probability of Antigenicity |
3.3. MHC Class II and I Epitopes
RANKPEP server used LACK, CPB, and KMP-11 proteins to anticipate MHC class II binding epitope (Tables 2). The MHC class I binding epitope anticipation was used by the server IEDB (Table 3).
Table 2. Details of MHC-II epitopes
| Protein Name | Start Position | Allele | Sequence | Score |
| CPB | 241 | DRB 0101 | YVSMESSER | 20.861 |
| 303 | DRB 0401 | YWVIKNSWG | 19.943 | |
| 116 | DRB 0101 | YRKARADLS | 18.08 | |
| 104 | DRB 0101 | YFAAAKQHA | 17.994 | |
| 105 | DRB 0401 | FAAAKQHAG | 17.737 | |
| 251 | DRB 1101 | MAAWLAKNG | 14.691 | |
| 163 | DRB 1101 | WAVAGHKLV | 10.425 | |
| KMP-11 | 70 | DRB 1101 | SEHFKQKFA | 17.535 |
| 77 | DRB 0101 | FAELLEQQK | 12.767 | |
| 48 | DRB 0401 | YEKFERMIK | 11.919 | |
| 5 | DRB 0101 | YEEFSAKLD | 9.643 | |
| LACK | 248 | DRB 0101 | FWMCVATER | 23.317 |
| 149 | DRB 0401 | HEDWVSSIC | 20.452 | |
| 41 | DRB 1101 | WKANPDRHS | 18.624 | |
| 26 | DRB 0101 | YIKVVLTSR | 18.44 | |
| 85 | DRB 1101 | WDRSIRMWD | 16.17 | |
| 10 | DRB 0401 | HRGWVTSLA | 15.115 |
Table 3. Details of MHC-I epitopes
| Protein Name | Start Position | Allele | Sequence | Score |
| CPB | 339 | HLA-A*01:01 | HVSQSPTPY | 0.41 |
| 265 | HLA-A*01:01 | VDASSFMSY | 0.35 | |
| 170 | HLA-B*07:02 | LVRLSEQQL | 0.28 | |
| 90 | HLA-A*01:01 | SEAEFAARY | 0.23 | |
| 73 | HLA-B*07:02 | QARNPHARF | 0.23 | |
| 118 | HLA-B*07:02 | KARADLSAV | 0.19 | |
| KMP--11 | 40 | HLA-A*01:01 | LSPEMKEHY | 0.35 |
| 69 | HLA-A*01:01 | HSEHFKQKF | 0.24 | |
| 34 | HLA-B*07:02 | KPDESTLSP | 0.12 | |
| 81 | HLA-A*01:01 | LEQQKAAQY | 0.11 | |
| LACK | 287 | HLA-A*01:01 | WSADGNTLY | 0.96 |
| 253 | HLA-A*01:01 | ATERSLSVY | 0.95 | |
| 159 | HLA-B*07:02 | SPSLEHPIV | 0.55 | |
| 188 | HLA-A*01:01 | RTLKGHSNY | 0.35 | |
| 19 | HLA-B*07:02 | CPQQAGSYI | 0.29 | |
| 164 | HLA-B*07:02 | HPIVVSGSW | 0.29 | |
| 42 | HLA-B*07:02 | KANPDRHSV | 0.28 |
3.4. Prediction of Interferon-gamma Induced Epitopes and B Cell Epitopes
The IFN epitope server was used to find the vaccine's IFN-gamma-inducing epitopes.
IFN gamma-inducing epitopes were categorized into 4 different epitopes. (Table 4).
BCpred server selected B-cell epitopes of three selected proteins. The results are shown in Table 5.
Table 4. IFN epitope server identified IFN-gamma-inducing epitopes in the ultimate vaccination design.
| Protein | Start–end position | IFN gamma score | Sequence |
| CPB | 104-124 | 1.6909292 | YFAAAKQHAGQHYRKARADLS |
| 241-260 | 2 | YVSMESSERVMAAWLAKNGP | |
| KMP-11 | 48-78 | 4.3950929 | YEKFERMIKEHTEKFNKKMHEHSEHFKQKF |
| LACK | 26-49 | 3.3236104 | YIKVVLTSRDGTAISWKANPDRHS |
Table 5. Details of B-cell epitopes
| Protein Name | Start Position | Sequence | Score |
| CPB | 209 | FTEKSYPYVSGNGDVPECSN | 0.988 |
| 300 | EVPYWVIKNSWGKDWGEKGY | 0.985 | |
| 329 | CLLTGYPVSVHVSQSPTPYL | 0.978 | |
| 126 | VPDAVDWREKGAVTPVKNQG | 0.977 | |
| KMP-11 | 31 | FADKPDESTLSPEMKEHYEK | 0.977 |
| LACK | 169 | SGSWDNTIKVWNVNGGKCER | 0.973 |
| 38 | AISWKANPDRHSVDSDYGLP | 0.931 | |
| 191 | KGHSNYVSTVTVSPDGSLCA | 0.898 |
3.5. Prediction of CTL Epitopes
The CTLpred server detected high-ranking CTL epitopes.
When the MHC-I binding data were examined, researchers analyzed the parts that overlapped with the CTLPred results. Furthermore, CTL epitopes derived from frequent sites were used. (Table 6).
Table 6. Predicted CPB, KMP-11, and LACK protein CTL epitope.
| Protein Name | Start Position | Sequence | Score |
| CPB | 63 | ERNLELMRE | 1.000 |
| 83 | ITKFFDLSE | 1.000 | |
| 115 | HYRKARADL | 1.000 | |
| KMP-11 | 60 | EKFNKKMHE | 0.990 |
| LACK | 25 | SYIKVVLTS | 1.000 |
| 104 | LKHTKDVLA | 0.990 |
3.6. Selection of Final Epitopes and Development of a Vaccine with Many Epitopes
According to high-grade and common MHC-I, MHC-II, CTL, B cell, INF-gamma epitopes, 8 sections of 3 antigenic proteins were selected as the final region (Table 7). The GSGSGS linker fused selected epitopes of each protein. RpfE and RpfB were also combined with both ends of the vaccine as adjuvants. The final structure of the vaccine consists of 518 amino acid residues, shown in Figure 1.
Table 7. The eight final epitope segments were selected based on the findings of the various servers from three antigens
| Protein Name | Start–end position | Sequence |
| CPB | 63-92 | ERNLELMREHQARNPHARFGITKFFDLSEA |
| 104-124 | YFAAAKQHAGQHYRKARADLS | |
| 219-260 | GNGDVPECSNSSELAPGARIDGYVSMESSERVMAAWLAKNGP | |
| 300-345 | EVPYWVIKNSWGKDWGEKGYVRVTMGVNACLLTGYPVSVHVSQSPT | |
| KMP-11 | 48-78 | YEKFERMIKEHTEKFNKKMHEHSEHFKQKFA |
| LACK | 26-49 | YIKVVLTSRDGTAISWKANPDRHS |
| 149-187 | HEDWVSSICFSPSLEHPIVVSGSWDNTIKVWNVNGGKCE | |
| 248-262 | FWMCVATERSLSVYD |

Figure 1. A representation of vaccine sequence arrangement.
3.7. Allergenicity and Antigenicity Evaluation
The result retrieved by the AllerTOP 2.0 server showed that the designed structure is non-allergenic.
As a result, the multiepitope vaccine has been created such that this will not generate allergy-specific IgE and inflammation.
The vaccine has a total antigenocytic probability of 0.8659 percent by Vaxijen at a minimum of 0.4 percent, indicating that it may induce effective T and B cell immune function.
3.8. Measurement of Physicochemical Parameters
According to our calculations, the protein's theoretical pI is 5.87, and its molecular weight is 55.05 kDa. The pI value is a measure of the protein's ph. Remainders with a total of 61 negative and 52 positive charges were found in this study. The anticipated half-lives of mammalian erythrocytes (in vitro), yeast (in vivo), and bacteria (in vitro) were 100, 20, and 10 hours, respectively (E. coli, in vivo). It was determined that the instability index (II) was 37.70. Stable protein is what we call this one. 61.58 was the aliphatic index value. The protein has a high aliphatic index, implying it can withstand a broad temperature. Aside from that, the vaccine construct's GRAVY value was -0.506. Negative GRAVY implies that the protein is hydrophilic and more capable of interacting with water molecules in the environment.
3.9. Secondary Structure Evaluation
Secondary structure prediction by GOR IV showed that the multi-epitope vaccine includes 24.13% α-helix (H), 20.66% extended strand (E), 0.00% beta-turn (T) and 55.21% random coil (C) (figure 2). The proportion of secondary structures in the multiepitope structure suggests that the designed vaccine is likely to be able to form antigenic epitopes.

 |
 |
Figure 2. The predicted secondary structure of the multiepitope vaccines using GOR IV software. H: Alpha helix (blue), E: Extended strand (red), T: Beta-turn (green) and C: Random coil (yellow)
3.10. Homogeneity Modeling
The I-TASSER server was used to create the final vaccine's 3D model, and the best five approaches rely upon C score were presented. The C score ranges from [-5 to 2], and the greater the C score, the more serious the problem, the more confident the model is. Model 3 had the best C-score (-1.76) and was chosen for more study. (Figure 3a).
3.11. Refinement of the Structure of the Third Model
Selected models were refined by applying GalaxyRefine. Five improved 3D models were released to the GalaxyRefine service. Furthermore, the best model was obtained based on the Z score and the overall quality coefficient of a high-quality three-dimensional model (Figure 3b).

Figure 3. The refined 3D model of the multiepitope peptide vaccine. A) The 3D structure of the designed vaccine was determined by homology modeling using I-TASSER server, and B) was refined by GalaxyRefine and 3Drefine servers.
3.12. Validation of the Refined Third Structure
Many virtual validating techniques were utilized to check the validity of the revised model. These approaches included: Ramachandran design; ERRAT; ProSA; and verify-3D.
For example, 337 (79.858 %), 56 (13.270 percent), and 29 (6.872 %) of the original model residues were found in the preferred, permitted, and outlier areas, respectively (Figure 4a).
393 (93.128 %), 18 (4.265 %), and 11 (2.607 percent) were the new ratios after the refining run (Figure 4b).
It seems from the findings that most of the amino acids have been moved inside the permitted range throughout the refining run, according to the data.

Figure 4. The validation of 3D protein model, using Ramachandran plot. a) The initial model b) The refined model
ProSA-web server assessments were also included in our research to further confirm the quality of the 3D model before and after refining. The original model's ProSA Z-score was -1.73, while the improved model's Z-score was -3.36 (Figure 5a and 5b, respectively). Structures of comparable size to natural proteins have been included in this model, as seen in Figure 5. It was also shown that most residues had negative energy values when plotted on a graph based on energy. (Figure 6).

Figure 5. The z-Score graphs for the construct's 3D structure.
The first model's z-score is -1.73, which is beyond the range of natural protein structure, and b
After refining, a model's z-score is – 3.63, which is within the range of natural protein structure.
The z-Score graphic shows the z-scores of all experimentally determined polypeptide chains in PDB (dark blue) and X-ray crystallography (light blue) (light blue).
Results with a z-score of 10 are shown in the graph.
The protein's z score is shown by a huge black dot.
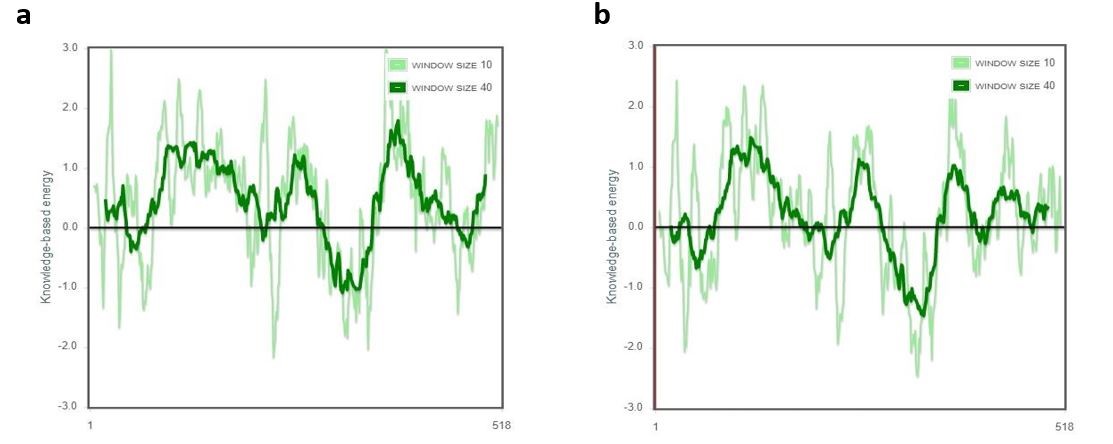
Figure 6. ProSA server energy charts of the original model (a) and revised model (b). For the improved model, the majority of the residues have negative numbers, as illustrated.
In addition, the quality of the modeled structure was verified using ERRAT. The results showed that the overall quality coefficient of the initial three-dimensional model was 58.5106 (Figure 7a). After refining processes, the ERRAT coefficient of the refined 3D model reached 73.4818 (Figure 7b).

Figure 7. The overall quality factor plot (ERRAT) of a the initial model is 58.5106, and b the final model after adjustment is 73.4818; areas of the 3D model that could be refused at the 99 percent confidence level are shown with grey lines, and zones that can be rejected at the 95 percent confidence level have been shown with black lines in the ERRAT graph. The total qualityof f value for excellent high-resolution structures is about 95% or above.
Furthermore, the Verify 3D score revealed that 53.86 percent of the residues in the original model had a mean score of 3D-1D score 0.2. (Figure 8a). The Verify 3D score was 58.69 percent after the refining procedure, showing that further residues were put in appropriate side chain settings. (Figure 8b).

Figure 8. The Verify-3D program evaluates the quality of 3D buildings. In the original model (a) and refined model (b), 53.86 percent and 58.69 percent of the residues, respectively, have a score of > 0.2.
3.13. In silico Cloning
The vaccine construct was developed by the Sequence Manipulation Suite server was backward translated and codon-optimized. The GenScript program was used to assess crucial gene sequence properties such as codon compatibility index (CAI), GC content, and codon frequency distribution in order to enable high-level protein production in E. coli hosts (CFD). CAI was the optimized nucleotide sequence 1, CAI> 0.80 is considered suitable for expression. The mean GC content of the designed vaccine sequence was 59.37%; the percentage of GC content in the range of 30-70% is desirable. The frequency distribution of the codon was 100 (CFD), while codons with values <30 appeared to be inhibited. Overall, these findings indicated that the optimized DNA sequence is clonable and expression-ready.
Leishmaniasis is a widespread parasitic disease that is prevalent in most regions of the globe. It may take many different forms, given the variety of pathogenic organisms. (61).
Leishmania major is one of the causes of occurring cutaneous leishmaniasis, which is more common in rural areas with poor health conditions and low economic status. Leishmania major can cause skin lesions. These lesions can spread and become painful. Tragically, even after healing, the lesion stays distorted, and the skin is injured in most instances. (62).
Various factors, including the unavailability of the vaccine, proper control of the vector, the cost of available drugs, the duration of treatment, and resistance to the parasite, all play a role in the inability to eradicate the disease completely. Therefore, the best strategy is to produce an effective vaccine that can stimulate and activate the body's immune system against the parasite (63, 64).
Basic studies are needed to identify a safe and effective vaccine that can stimulate the immune response (65). Antigens such as CP and KMP-11 have been investigated as targets in vaccine design for combat Leishmania (66, 67). In addition, recombinant vaccines such as Leish111f and Leishmune have been developed as leishmaniasis vaccination alternatives (68). Yet, no ideal vaccination against the illness has been authorized for human treatment (13, 69).
Bioinformatics approaches are now effective for designing novel vaccines (70). Several important studies have demonstrated the benefits of vaccines that have been designed using bioinformatics methods and are currently used as an effective vaccine (e.g., studies on malaria, influenza, cancer, dengue, and multiple sclerosis) (19, 71-73).
Several bioinformatics and immunoinformatics techniques have been effectively applied in numerous biological disciplines. These tools reduce the time and cost required to identify the dominant immune epitopes of B and T cells while increasing the level of screening accuracy (74).
Research shows that Leishmania major infection in a resistant mouse model; it activates Th1 type CD4 cells. Th1 cells produce interferon-gamma, which promotes improvement and immunity against infection. The Th2 response is induced in BALB/c sensitive mice, and IL-4 is generated; however, IFN- production is decreased, and infected animals are susceptible to the illness. However, in the mouse model, the production of the Th1 response type is linked to therapy and protection, while the generation of the Th2 response type is linked to disease progression and mortality, but human leishmaniasis susceptibility and resistance are yet unknown (20, 75-78).
Overall, the recovery form of CL is associated with cells that generate IFN-, while the non-recovery form of CL is associated with a combination of Th1 and Th2 cytokines, with IL-4 and IL-10 being abundant (79-81). As a result, vaccinations targeting T and B cell epitopes seem to be more effective. As a result, T and B cell epitopes were investigated in this research.
Employing bioinformatics techniques, the antigens LACK, CPB, and KMP-11 were utilized to construct a multiepitope vaccination for L. major. In order to enhance the likelihood of discovering the greatest immunodominant epitopes, numerous servers were used to choose LACK, CPB, and KMP-11 antigens. MHC-I, MHC-II, CTL, B cell, and INF-gamma binding epitopes were compared, and epitopes with high scores and overlaps were chosen. In addition to predicting T and B cell epitopes, the presence of IFN-γ-induced epitopes in the structure of the ultimate vaccine was evaluated. Four epitopes were identified as IFN-γ-induced epitopes. Several cytokines, notably IFN-, have been demonstrated to be significant in parasite elimination. (82). Hence, epitopes capable of producing IFN-γ are important for CL vaccine design.
Adjuvants are added to the structure of multiepitope peptide vaccines to overcome the poor immunogenicity of the vaccine. Adjuvants play a crucial role in boosting the immune system (41, 83). Pathogen-associated molecular patterns (PAMPs) attach to Toll-like receptors (TLRs), which trigger innate immune responses. Two TLR 4 agonists, RpfB and RpfE, were utilized as adjuvants in this investigation. RpfB enhances Th1-type T cell immunological responses by directly binding to TLR4 and activating TLR4-dependent dendritic cells. RpfE interacts with DC to differentiate crude CD4 cells into Th1 and Th17 immune responses (62). As a result, using adjuvants to develop effective multiepitope vaccinations to prevent CL is a good idea.
Linkers are essential components of recombinant multiepitope vaccines and take part in functional structural vaccine development (79, 84). In this study, two types of linkers, namely EAAAK and GSGSGS, were used to join different parts of the multiepitope vaccine. The GSGSGS flexible linker was used to match functional domains that require domain interactions. EAAAK rigid linker creates space between domains (80, 81). In addition, these linkers provide stability and bioactivity to the peptide structure (83, 85). Accordingly, EAAAK was used to link RpfB and RpfE to the designed vaccine structure.
Bioinformatics tools evaluated the vaccine's physicochemical, immunological, and structural characteristics. According to the results of structural, immunological, and physicochemical properties, the vaccine designed in this study can be proposed as a suitable option for a vaccine.
Numerous factors, including CAI, CFD, and GC content, must be tuned to achieve high-level protein expression in E. coli. The findings of the optimized gene demonstrated that the intended vaccine could be efficiently produced in the E. coli host following analyzing all of the aforementioned parameters employing GenScript.
Using an immunoinformatics approach, we aimed to construct a multiepitope-based vaccination against CL in this work. Our created vaccine may be a decent choice for vaccination against Leishmania major based on computational findings, immunological and structural analyses, and physicochemical assessments. The recombinant structure can activate both humoral and cellular immune responses. The suggested vaccine, which uses a variety of epitopes and adjuvants and has acceptable physicochemical properties, is likely to elicit strong immune responses against CL. However, in vitro and in vivo testing are required to assess the effectiveness of multiple epitope vaccines.
The authors appreciate the Deputy of Research and Technology, Lorestan University of Medical Sciences, Khorramabad, Iran. This article is derived from the Master's thesis of the first author, Department of Biotechnology, School of Medicine, Lorestan University of Medical Sciences, Khorramabad, Iran.
The present study was approved by The Ethics Committee of Lorestan University of Medical Sciences (IR.LUMS.REC.1398.190).
This research was financially supported by Lorestan University of Medical Sciences, Khorramabad, Iran. Hereby the authors appreciate all the people who helped in this research.
Conflicts of Interest
The authors declare that there is no conflict of interest.
Received: 2021/12/25 | Accepted: 2022/06/4 | ePublished: 2022/08/8
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
Copyright Policy
Iranian Journal of Medical Microbiology by Farname is licensed under CC BY-NC 4.0![]()
![]()
![]()
